Żółto i niebiesko - barwnik elektrochromowy
Poniższy artykuł został opublikowany pierwotnie w czasopiśmie dla nauczycieli Chemia w Szkole (5/2017):

Według wielu specjalistów dzisiejsze czasy należałoby nazwać Wiekiem Informacji. Społeczeństwo, w którym informacja staje się towarem traktowanym jako szczególne dobro niematerialne - równoważne lub nawet cenniejsze od dóbr materialnych - nazywa się społeczeństwem informacyjnym [1]. Ważną jego cechą jest szybki rozwój usług związanych z sektorem 3P, czyli polegających na przesyłaniu, przetwarzaniu i przechowywaniu różnego rodzaju danych.
Cały ogrom informacji dostępnych dzisiaj dla prawie każdego musi być przetwarzany w odpowiedni sposób. Pomagają nam w tym specjalizowane urządzenia - dawniej mechaniczne (np. kieszonkowy kalkulator Curta z 1948 roku [2]), dziś elektroniczne (komputery, telefony komórkowe).
Interakcja urządzeń przetwarzających informację z człowiekiem wymaga jakiegoś sposobu komunikacji. Jednym z możliwych rozwiązań tego zagadnienia są wszelkiego rodzaju wyświetlacze, czyli urządzenia elektroniczne służące do wyświetlania informacji.
Znamy wiele typów konstrukcji wyświetlaczy wykorzystujących różnego rodzaju zjawiska fizyczne lub fizyko-chemiczne. Niewielki przegląd tego typu urządzeń przedstawia Fot.1.

Dawniej w sprzęcie pomiarowym do wyświetlania wyników pomiarów poza wskaźnikami elektromechanicznymi (wskazówkowymi) były dosyć powszechnie stosowane tzw. digitrony, zwane też lampami Nixie. Były to wyspecjalizowane lampy neonowe, w których pod wpływem odpowiednio wysokiego napięcia świecił wzbudzony gaz – zazwyczaj neon pod bardzo niskim ciśnieniem. Z racji odpowiedniego ukształtowania elektrod digitron mógł wyświetlać określony zestaw znaków, najczęściej cyfry od 0 do 9, czasem wraz z punktem dziesiętnym (Fot.1A). Wyświetlacze takie, mimo docenianych dzisiaj walorów estetycznych, miały kilka dosyć poważnych wad: zasada ich działania nie pozwalała na wyraźną miniaturyzację, a do ich zasilania było potrzebne stosunkowo wysokie napięcie (zwykle rzędu 100V). Inną konstrukcją wyświetlaczy, stosowaną w pewnych urządzeniach nawet dzisiaj, są próżniowe wyświetlacze fluorescencyjne (ang. Vacuum Fluorescent Display, VFD). Źródłem światła w tym przypadku jest odpowiednio ukształtowana warstwa luminoforu, bombardowana elektronami przyspieszanymi przez pole elektryczne w próżni (Fot.1B). Źródłem elektronów jest rozgrzewana przepływem prądu elektrycznego katoda wykonana z wolframu. Urządzenia tego typu mogły być już bardziej zminiaturyzowane, ale wymagały zwykle kilku napięć zasilających (np. osobne zasilanie żarnika i anod), co przekładało się na dosyć kłopotliwe sterowanie.
Dużo nowszym rozwiązaniem są siedmiosegmentowe wyświetlacze zbudowane z diod elektroluminescencyjnych LED zamkniętych w odpowiedniej obudowie (Fot.1C). Są one zasilane napięciem rzędu pojedynczych woltów, a trwałością zdecydowanie przewyższają dawniejsze rozwiązania. Jeszcze bardziej energooszczędne są wyświetlacze ciekłokrystaliczne, jakie są dziś stosowane najczęściej w sprzęcie elektronicznym powszechnego użytku (Fot.1D).
Oczywiście nie są to wszystkie znane technologie wyświetlaczy. Od pewnego czasu obserwuje się coraz większe zainteresowanie rozwiązaniami typu e-papieru, czyli rodzaju wyświetlacza, który swoimi wymiarami i właściwościami mechanicznymi stara się naśladować papier. Trwają badania nad technologiami przydatnymi w tym celu – bierze się pod uwagę pomysły oparte na wyświetlaczach ciekłokrystalicznych, plazmowych, elektrochemicznych i innych.
Wydaje się, że można w tym kierunku wykorzystać zjawisko elektrochromizmu, tj. zmiany barwy pewnych substancji chemicznych w zależności od przepływu prądu elektrycznego.
Jak się Szanowny Czytelnik pewnie domyśla, oczywiście nie poprzestaniemy na rozważaniach teoretycznych - dokonamy odpowiedniej syntezy, a następnie przekonamy się naocznie o elektrochromowych właściwościach pewnej ciekawej substancji.

Pruski czy Turnbulla?
Interesująca nas substancja ma nieco zawiły rodowód. Z chemicznego punktu widzenia należy ją nazwać heksacyjanożelazianem(II) potasu żelaza(III). Najczęściej przytaczany wzór sumaryczny tego związku ma postać KFe[Fe(CN)6].
Jak widać, jest to nieorganiczny związek chemiczny z grupy kompleksowych soli mieszanych. Zawiera kationy potasu i żelaza(III) oraz anion heksacyjanożelazianowy(II), którego wzór strukturalny jest przedstawiony na Rys.1.
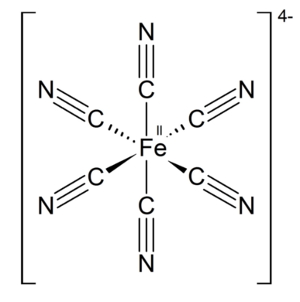
Omawiany związek chemiczny wykazuje piękne niebieskie zabarwienie - można je wyjaśnić absorpcją światła powodującą przeniesienie elektronu pomiędzy atomami żelaza istniejącymi na różnych stopniach utlenienia.
Po raz pierwszy ten niebieski barwnik otrzymał w 1709 roku Johann Konrad Dippel.
Dawniej rozróżniano dwa podobne środki barwiące: błękit pruski i błękit Turnbulla. Pierwsza nazwa wzięła swój początek od faktu, że błękit został zastosowany przy barwieniu mundurów pruskich żołnierzy, co miało odróżniać je od zielonych strojów stosowanych w armii rosyjskiej i niebieskich (o innym odcieniu) w francuskiej oraz bawarskiej [3].
W wyniku badań struktury wspomnianych barwników okazało się, że pod względem chemicznym są one identyczne [4]. Tradycyjnie przyjmuje się jednak, że otrzymuje się je w wyniku odmiennych reakcji chemicznych. Tak więc błękit pruski powstaje w wyniku reakcji kationów żelaza(III) i anionów heksacyjanożelazianowych(II), natomiast błękit Turnbulla w wyniku reakcji kationów żelaza(II) i anionów heksacyjanożelazianowych(III). Obie reakcje muszą oczywiście zachodzić w obecności kationów potasu.
Nikt nam jednak nie każe wierzyć na słowo, więc powinniśmy to sprawdzić.
Myślę, że w tym miejscu należy wyraźnie odróżnić od siebie odpowiednie heksacyjanożelaziany. Heksacyjanożelazian(II) potasu K4[Fe(CN)6], nazywany też żelazocyjankiem potasu lub solą Gmelina jest cytrynowożółtym ciałem krystalicznym (Fot.2A). Heksacyjanożelazian(III) potasu K3[Fe(CN)6] (żelazicyjanek potasu), ma natomiast postać pięknych, czerwonych kryształów (Fot.2B).

Przygotujmy roztwory soli żelaza dwuwartościowego i trójwartościowego. Tu pojawia się pewna trudność, ponieważ kationy żelaza(II) w wielu związkach i ich roztworach są podatne na utlenianie do żelaza(III). Stosunkowo mało kłopotliwym źródłem potrzebnych jonów dwuwartościowych jest tzw. sól Mohra, czyli sól podwójna kwasu siarkowego(VI) oraz amonu i żelaza(II) o wzorze (NH4)2Fe(SO4)2. Źródłem jonów żelaza(III) może być natomiast np. chlorek żelaza(III) FeCl3. Roztwory obu soli należy rozlać do osobnych probówek (Fot.3A i B).

Po dodaniu do heksacyjanożelazianu(III) do soli żelaza(II) oraz heksacyjanożelazianu(II) do soli żelaza(III) w obu przypadkach uzyskano niebieskie zabarwienie próbek. W przypadku C było to spowodowane powstaniem błękitu Turnbulla, zaś w przypadku D błękitu pruskiego.
Oczywiście, jak już wspomniano, powyższe rozróżnienie ma charakter raczej zwyczajowy lub techniczny. Trzeba jednak zaznaczyć, że odcień pigmentu uzyskanego obiema metodami może się nieco różnić – jest to jednak spowodowane jedynie odmiennymi zanieczyszczeniami i domieszkami.
Zauważmy, że odpowiednie heksacyjanożelaziany pozwalają na wykrywanie i rozróżnianie (jako, że tworzą błękit pruski/Turnbulla jedynie w podanych kombinacjach) soli żelaza dwuwartościowego i trójwartościowego.
Sprawa nazewnictwa komplikuje się jeszcze bardziej, ponieważ przy niestechiometrycznych proporcjach reagentów powstaje niebieski barwnik opisywany często wzorem FeIII4[FeII(CN)6]3. Jest on jednak w odróżnieniu od poprzednio wspomnianych pigmentów z racji swoich właściwości nazywany nierozpuszczalnym błękitem pruskim [5].
By zapoznać się z interesującymi właściwościami barwników zbliżonych do błękitu pruskiego musimy dokonać odpowiedniej syntezy.
Hodowla warstwy
Warstwę barwnika wytworzymy na odpowiednim podłożu.
W celu przeprowadzenia proponowanych doświadczeń potrzebujemy biernej chemicznie elektrody, która dodatkowo byłaby w jak największym stopniu przezroczysta. Wymagania te spełnia szkło pokryte jednostronnie przejrzystą warstwą półprzewodnika np. wykonaną z mieszanego tlenku indu i cyny (ang. indium tin oxide, ITO). Tego typu podłoże wykorzystałem z powodzeniem także w eksperymentalnym fotoogniwie uczulanym barwnikiem antocyjanowym pochodzącym z malin [6].
Musimy sprawdzić, która z powierzchni szkła jest pokryta warstwą półprzewodnika. Najłatwiej zrobić to za pomocą miernika rezystancji (Fot.4). Ja wykorzystałem płytkę szkła o wymiarach 5cm na 2cm.

Jak widać, pomiar wykazał wartość 82,5Ω przy elektrodach pomiarowych odległych od siebie o 2,7 cm. Przewodność warstwy jest więc dużo mniejsza niż w przypadku metali, ale wystarczy w zupełności.
Szklaną płytkę trzeba dokładnie umyć i odtłuścić - najpierw w wodzie z detergentem, a następnie w acetonie.
Do dalszych prac większość powierzchni przewodzącej płytki musimy pokryć warstwą izolacyjną. W wielu publikacjach zaleca się tutaj stosowanie warstwy teflonu, parafiny lub innych metod. W toku moich doświadczeń stwierdziłem jednak, że całkiem wystarczająca jest tutaj przejrzysta biurowa taśma samoprzylepna.
Powierzchnię przewodzącą należy pokryć taśmą samoprzylepną według Fot.5.

W warstwie tworzywa sztucznego, z jakiego jest wykonana taśma należy naciąć ostrym narzędziem, a następnie odsłonić fragmenty powierzchni a i b. Pierwsze stanie się miejscem osadzania barwnika, zaś do drugiego podłączymy jeden z biegunów źródła zasilania. Pole a powinno mieć powierzchnię 1cm2, tj. być kwadratem o boku równym 1cm - przydatne jest przy tym użycie papieru milimetrowego podłożonego pod szkło. Wymiary pola b nie są krytyczne.
Druga elektroda musi także charakteryzować się wysoką biernością chemiczną. Z tego powodu najlepsza byłaby tu elektroda platynowa, lecz w przypadku jej braku można się posłużyć elektrodą grafitową. Obie elektrody należy umocować w sposób zapewniający ich usytuowania w bliskiej wzajemnej odległości, ale tak by ich powierzchnie przewodzące nie mogły się zetknąć. Dosyć dobrym rozwiązaniem jest zastosowanie w tym celu zacisków krokodylkowych, jak widać to na Fot.6.

Aby przygotować potrzebny elektrolit musimy najpierw sporządzić trzy roztwory:
- A – 0,05M HCl (2cm3 stężonego kwasu w 100cm3 wody destylowanej)
- B – 0,05M K3[Fe(CN)6] (1,65g soli w 100cm3 wody destylowanej)
- C – 0,05M FeCl3 (1,35g soli w 100cm3 wody destylowanej z dodatkiem kilku kropli HCl) [7]
Dodatek kwasu w roztworze C ma za zadanie zahamować hydrolizę chlorku żelaza(III).
Przy sporządzaniu roztworów pamiętajmy, że stężony kwas chlorowodorowy jest żrący, a ulatniający się z niego gazowy chlorowodór ma działanie drażniące. O ile heksacyjanożelazian(III) nie jest silnie toksyczny, to w mieszaninie z mocnymi kwasami (szczególnie po podgrzaniu) rozkłada się z wydzieleniem bardzo trującego cyjanowodoru HCN. Należy zachować ostrożność.
Elektrolit przygotowujemy poprzez zmieszanie roztworów w podanej kolejności, w stosunku objętościowym A:B:C = 1:2:2. Zauważmy, że nie dochodzi wtedy do powstania niebieskiego zabarwienia – nie ma w tym żadnej zagadki, ponieważ zmieszaliśmy heksacyjanożelazian(III) z solą żelaza(III). Powstały w ten sposób elektrolit ma postać brunatnoczerwonej cieczy (Fot.7).

Elektrolit należy sporządzać bezpośrednio przed dalszymi etapami doświadczenia, ponieważ nie nadaje się on do dłuższego przechowywania.
Zestaw elektrod należy umieścić w elektrolicie – odsłonięte rejony warstwy przewodzącej płytki szklanej muszą być bezpośrednio przed procesem dodatkowo dokładnie oczyszczone i odtłuszczone. Trzeba przy tym zadbać, aby metaliczne styki doprowadzające prąd elektryczny do elektrod nie wchodziły w kontakt z elektrolitem, a kwadratowe pole wycięte w warstwie izolacyjnej było w całości zanurzone (Fot.8).

Przystępując do prowadzenia elektrolizy należy pamiętać, że katodą powinna być tutaj płytka szklana.
Elektrolizę należy prowadzić w temperaturze pokojowej, przy niewielkim napięciu - w moim przypadku było to niewiele powyżej 1V. Gęstość prądu podczas elektrolizy musi mieć wartość zbliżoną do 40 μA/cm2. Jako, że aktywna (odkryta, zanurzona w elektrolicie) powierzchnia katody wynosi właśnie 1cm2, to natężenie prądu podczas elektrolizy powinno wynieść około 40μA . Stosując odpowiedni opór włączony szeregowo z elektrolizerem można dosyć dokładnie wyregulować natężenie prądu, tak jak w przypadku moich doświadczeń (Fot.9).

Wraz z upływem czasu elektrolizy na podłożu szklanym (a raczej na warstwie przewodzącej) osadza się coraz grubsza warstwa niebieskiego barwnika. Warstwy zbyt cienkie mają niewyraźną barwę, zaś zbyt grube nie wykazują dobrego kontaktu elektrycznego z podłożem. Warto więc w tym przypadku poeksperymentować z czasem trwania procesu.
Po upływie założonego czasu należy odłączyć zasilanie, wyciągnąć elektrody z cieczy, a następnie wypłukać je w wodzie destylowanej.
W przypadku moich doświadczeń prowadziłem proces w czasie 90 sekund, co skutkowało powstaniem wyraźnie niebieskiej, lecz w dalszym ciągu przejrzystej warstwy (Fot.10).

Warstwa barwnika jest delikatna – nie należy jej dotykać ani wycierać, a jedynie przepłukiwać powolnym strumieniem wody destylowanej. Można teraz ostrożnie usunąć warstwę izolacyjną (Fot.11). Płytkę do czasu dalszych manipulacji najlepiej przechowywać w stanie zwilżenia wodą destylowaną.

Mając gotową płytkę z barwnym polem możemy przejść do właściwych doświadczeń.
Zmiany barwy
Musimy sporządzić kolejny elektrolit. Tym razem będzie to 1M roztwór chlorku potasu KCl w wodzie (7,44g KCl w 100cm3 wody) zakwaszony paroma kroplami HCl.
W elektrolicie zanurzamy przygotowaną elektrodę z barwnikiem, a także elektrodę grafitową (Fot.12).

Jako źródło napięcia w tym przypadku zastosujemy suche ogniwo Leclanchégo, np. typu R6 (tzw. paluszek). W opisanym przypadku wymagana jest konieczność wygodnej zmiany biegunowości elektrod oraz możliwość zwarcia ich ze sobą. Można to oczywiście robić ręcznie łącząc odpowiednio przewody, ale dużo lepszym rozwiązaniem jest wykorzystanie dwóch dwuobwodowych przełączników zestawionych według schematu zamieszczonego na Rys.2.
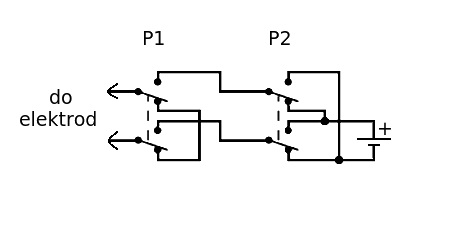
Jak widać przełącznik P2 umożliwia zmianę kierunku przepływu prądu w dalszej części obwodu (a więc także w elektrolizerze), natomiast przełącznik P1 pozwala na doprowadzenie prądu do elektrod lub ich zwarcie ze sobą.
Zbudowany według powyższego opisu zestaw przełączników przedstawia Fot.13.

Ważne jest, aby po zanurzeniu elektrod w elektrolicie zacząć od ich zwarcia. Barwne pole jest wtedy oczywiście niebieskie (Fot.14A).

Następnie należy podłączyć elektrodę szklaną do ujemnego bieguna źródła zasilania, zaś grafitową do dodatniego. Możemy wtedy zaobserwować interesujące zjawisko: niebieskie pole w bardzo krótkim czasie praktycznie znika. Jedynie patrząc pod pewnym kątem można zaobserwować, że warstwa pigmentu w dalszym ciągu pozostała na swoim miejscu, ale stała się praktycznie bezbarwna (Fot.14B).
Po zwarciu elektrod ze sobą pole z powrotem staje się niebieskie.
Opisaną sekwencję polaryzowania elektrody szklanej ujemnie względem grafitowej i zwierania elektrod należy na początku powtórzyć kilkanaście razy. Umożliwia to dalszą, stabilną pracę układu elektrochemicznego i przedłuża jego żywotność.
Kolejną wartą wypróbowania rzeczą jest zmiana polaryzacji elektrod: elektrodę szklaną podłączamy wtedy do dodatniego bieguna źródła zasilania, zaś elektrodę grafitową do ujemnego. W takim przypadku barwne pole staje się wyraźnie żółte (Fot.14C). Zwarcie elektrod przywraca ponownie barwę niebieską.
Zmieniając polaryzację elektrod, a także zwierając je ze sobą można wpływać na kolor barwnego pola. Najlepiej jest, aby pomiędzy cyklami zmiany polaryzacji występował moment zwarcia elektrod.
Tak przygotowana elektroda z barwnikiem jest dosyć trwała nawet w stanie suchym, ale dobrze jest ją przechowywać zanurzoną w elektrolicie chlorkowo-potasowym, oczywiście w naczyniu z ciemnego szkła.
W razie potrzeby warstwę barwnika można usunąć poprzez płukanie płytki w stężonej wodzie amoniakalnej NH3(aq).
Wyjaśnienie
W czasie elektrolitycznej hodowli barwnika dochodzi do powstania wspomnianego wcześniej nierozpuszczalnego błękitu pruskiego Fe4III[FeII(CN)6]3, który dzięki oddziaływaniom elektrostatycznym formuje jednorodną warstwę o zabarwieniu niebieskim.
Kiedy elektroda szklana jest katodą, to odbywa się na niej proces redukcji barwnika w obecności kationów potasowych K+ według równania:
Powstały w ten sposób związek o wzorze K4Fe4II[FeII(CN)6]3 jest nazywany solą Everitta i w postaci cienkiej warstwy jest praktycznie bezbarwny [8]. Powoduje to oczywiście zanik barwnego pola.
Inaczej będzie, gdy elektroda szklana stanie się anodą. W takim przypadku błękit zostanie utleniony w obecności anionów chlorkowych Cl-:
Otrzymany w ten sposób związek o wzorze Fe4III[FeIII(CN)6]3Cl3 w postaci cienkiej warstwy ma zabarwienie żółte i jest nazywany żółcienią pruską (ang. prussian yellow).
Dosyć ważne jest przynajmniej kilkukrotne powtórzenie na początku cyklu polaryzacji elektrody szklanej jako katody i zwarcia elektrod. W tym czasie struktura barwnika traci około jednej czwartej kationów żelaza(III) dzięki wymianie na kationy potasu. Elektroneutralność zostaje zachowana, a warstwa nabiera właściwości rozpuszczalnego błękitu pruskiego [9].
Jak widać, z łatwością można sobie wyobrazić budowę wyświetlacza wykorzystującego tego typu związki chemiczne i umożliwiającego prezentację różnego rodzaju informacji, np. dzięki zastosowaniu przejrzystych elektrod i pól barwnych o odpowiednim kształcie.
Literatura:
- [1] Goban-Klas T., Sienkiewicz P., Społeczeństwo informacyjne: Szanse, zagrożenia, wyzwania, Wydawnictwo Fundacji Postępu Telekomunikacji, Kraków, 1999 powrót
- [2] C. Stoll, The Curious History of the First Pocket Calculator, Scientific American, 290 (1), 2004, str. 92–99 powrót
- [3] Pastoreau M., Niebieski - historia koloru, Oficyna Naukowa, Warszawa, 2013 powrót
- [4] Hansen L.D., Litchman W.M., Daub G.H., Turnbull’s blue and Prussian blue: KFe(III)[Fe(II)(CN)6], Journal of Chemical Education, 46 (1), 1969, str. 46 powrót
- [5] Izatt R.M., Watt G.D., Bartholomew C.H., Christensen J.J., Calorimetric study of Prussian blue and Turnbull’s blue formation, Inorganic Chemistry, 9 (9), 1970, str. 2019–2021 powrót
- [6] Ples M., Fotoogniwo uczulane barwnikiem. Czego możemy nauczyć się od natury?, Chemia w Szkole, 3 (2017), Agencja AS Józef Szewczyk, str. 6-12 powrót
- [7] Marmon J., Lisensky G., Electrochromic Prussian Blue Thin Films, w serwisie: http://education.mrsec.wisc.edu/, dostępne online: http://education.mrsec.wisc.edu/291.htm [dostęp 26.09.2017] powrót
- [8] Kraft A., On The History Of Prussian Blue: Thomas Everitt (1805-1845) And Everitt’s Salt, Bulletin for the History of Chemistry, 39 (1), 2014 powrót
- [9] García-Jareño J.J., Benito D., Navarro-Laboulais J., Vicente F., Electrochemical Behavior of Electrodeposited Prussian Blue Films on ITO Electrodes - An Attractive Laboratory Experience, Journal of Chemical Education, 75 (7), 1998 powrót
Wszystkie fotografie i rysunki zostały wykonane przez autora
Uzupełnienie autora
Poniżej przedstawiam filmik obrazujący zmiany barwy cienkiej warstwy opisywanej w artykule substancji.
Marek Ples