Lofina - wielka synteza
Poniższy artykuł został opublikowany pierwotnie w czasopiśmie dla nauczycieli Chemia w Szkole (6/2022):

Mimo że chemia – jak i nauka jako całość – jest fascynującym elementem naszego życia, to w moim mniemaniu nie będzie błędne stwierdzenie, iż istnieją pewne grupy reakcji chemicznych, które zawsze cieszą się szczególnie wielkim zainteresowaniem uczniów i nauczycieli. Dzieje się tak często nie tylko ze względu na niewątpliwą wartość dydaktyczno-naukową, ale też wspaniały efekt wizualny. Do tego typu procesów należą reakcje chemiluminescencyjne, czyli takie, w których dochodzi do emisji promieniowania elektromagnetycznego z zakresu światła widzialnego na drodze innej niż termiczna. Chociaż takie reakcje wydają się być czymś rzadko spotykanym, to obok stosunkowo drogich, trudno dostępnych lub skomplikowanych w syntezie substancji takich jak np. lucyferyna robaczków świętojańskich Lampyris noctiluca (i innych chrząszczy, m.in. iskrzyków Phausis splendidula, przyp. aut.) C11H8N2O3S2, lucygenina C28H22N4O6 i tetrakis(dimetyloamino)etylen C10H24N4 można je zaobserwować także w przypadku dużo łatwiejszych do zdobycia chemikaliów. Do zimnego świecenia zmusić możemy białą odmianę alotropową fosforu P, metaliczny sód Na i potas K, formę singletową tlenu 1O2, polifenole wchodzące w skład zielonej herbaty, niektóre związki Grignarda i krzemoorganiczne (wytworzony ze zwykłego piasku siloksen Wöhlera Si6O3H6), a nawet łatwo dostępny w aptekach manganian(VII) potasu KMnO4 [1][2][3][4][5]. Jednym z chemiluminoforów jest także będąca przedmiotem niniejszego opracowania lofina C21H16N2.

Warto wspomnieć, że z pierwszą syntezą lofiny można powiązać bardzo interesujące wydarzenia i postaci historyczne. Dokonał tego w drugiej połowie XIX wieku nasz rodak, a jednocześnie znakomity chemik-organik Bronisław Radziszewski. Pierwsza opracowana przez niego metoda syntezy lofiny była możliwa przy wykorzystaniu innego związku chemicznego, czyli tribenzylidenodiiminy, nazywanej też hydrobenzamidem. Tą substancję opisał z kolei jako pierwszy Aleksander Borodin. Tutaj możemy się zdziwić, ponieważ jego postać kojarzymy raczej z historią muzyki, a nie chemii. Był kompozytorem i wraz z Bałakirewem, Cui, Musorgskim i Rimskim-Korsakowem tworzył Potężną Gromadkę - grupę sławnych twórców muzycznych nawiązujących do tradycyjnej rosyjskiej muzyki ludowej. Komponował symfonie, poematy symfoniczne, fantazje i kwartety smyczkowe. Co ciekawe, Borodin traktował muzykę jako swoje uboczne zajęcie (z którego jest jednak do dziś powszechnie znany), natomiast jego zawodową i największą pasją zawsze była chemia.
W jednym z dawniejszych numerów Chemii w Szkole opisałem moje próby (udane, przyp. aut.) przeprowadzenia historycznie pierwszej metody syntezy lofiny zaproponowanej przez Radziszewskiego [6]. Metodę tę wybrałem, ponieważ jako głównego surowca wykorzystuje ona łatwo dostępny i tani aldehyd benzoesowy, podczas gdy nowsza oraz bardziej wydajna metoda wielkolaboratoryjna wymaga użycia trudniejszego do zdobycia benzilu. Wadą metody historycznej jest jednak jej bardzo niska wydajność. Zastanawiając się oraz eksperymentując nad zwiększeniem wydajności procesu rozpoczynającego się od aldehydu (i innych łatwych do zdobycia substancji) doszedłem do wniosku, że jest to możliwe. Będzie to wieloetapowa synteza (aldehyd benzoesowy → benzoina → benzil → lofina), co dodatkowo pozwoli nam w prosty, a jednocześnie stosunkowo bezpieczny sposób zapoznać się ze szczegółami tego rodzaju prac. Nie zwlekając więc dłużej przystąpmy do pierwszego etapu syntezy. Pamiętajmy przy tym, że wszystkie prace musimy wykonywać stosując odpowiednie środki ochrony osobistej.
Etap I – od aldehydu benzoesowego do benzoiny
Rozpoczynając nasze prace musimy zgromadzić substancje takie jak:
- aldehyd benzoesowy C6H5CHO,
- etanol C2H5OH 95% (spirytus rektyfikowany),
- tiamina C12H17N4OS·HCl (witamina B1 w postaci chlorowodorku),
- wodorotlenek sodu NaOH, roztwór wodny 10% (na podstawie [7], zmodyfikowano).
Aldehyd benzoesowy jest przejrzystą cieczą, o silnym zapachu migdałów. W rzeczywistości jest odwrotnie – to migdały pachną aldehydem, którego niewielkie ilości w nich występują. Zasadniczo do doświadczenia można by użyć aldehydu benzoesowego otrzymanego naturalnie z nasion śliwy migdał Prunus dulcis, częściej nazywanej migdałowcem. Cena takiej substancji byłaby jednak bardzo wysoka, dlatego łatwiej wykorzystać aldehyd wytworzony syntetycznie, jak i ja zresztą zrobiłem.
Aldehyd benzoesowy łatwo się utlenia (szczególnie na świetle), dlatego należy go chronić przed dostępem powietrza. Wraz z postępem tego procesu ciecz staje się najpierw żółtawa, a potem brunatna. Do naszego doświadczenia jest potrzebny świeży aldehyd, to znaczy bezbarwny lub co najwyżej z delikatnym żółtym odcieniem. Silnie utlenione próbki substancji nie nadają się i należałoby je wcześniej przedestylować, by odzyskać pozbawiony produktów utleniania aldehyd. Aldehyd może być szkodliwy dla naszego organizmu.
Tiamina C12H17N4OS (później nazwana witaminą B1) to heterocykliczny związek chemiczny, złożony z pierścieni tiazolowego i pirymidynowego, połączonych mostkiem metylenowym. Substancję tę wyodrębnił w 1911 roku z otrębów ryżowych Kazimierz Funk, po czym zaproponował dla niej i podobnych jej niezbędnych ludziom do życia substancji nazwę witamina. Strukturę jej cząsteczki przedstawia Rys.1.
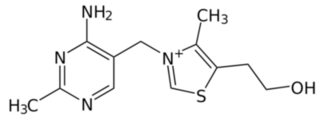
Tiamina ma w normalnych warunkach postać białego ciała drobnokrystalicznego (Fot.1). Jest rozpuszczalna w wodzie i odgrywa zasadniczą rolę w procesach oddychania komórkowego, głównie w przemianie węglowodanów i wchodzi w skład koenzymu karboksylazy. Wzmaga aktywność acetylocholiny C7H16NO2, hamuje esterazę cholinową, działa synergicznie z tyroksyną C15H11I4NO4 i insuliną, pobudza też wydzielanie hormonów gonadotropowych. Tiamina przyspiesza gojenie się ran i wykazuje działanie uśmierzające ból. Z badań wynika, że dzienne zapotrzebowanie dorosłego człowieka na witaminę B1 wynosi 1,1 mg dla kobiet i 1,2 mg dla mężczyzn [8].

Do skutków niedoboru tej substancji w naszym organizmie należą między innymi zaburzenia czynności centralnego układu nerwowego (osłabienie, zmęczenie, oczopląs, zaburzenia pamięci, koncentracji, depresja), niewydolność krążenia (przyspieszona akcja serca, powiększenie wymiarów serca, obrzęki kończyn górnych i dolnych), zaburzenia ze strony przewodu pokarmowego (utrata łaknienia, nudności, wymioty, biegunka, ból z okolic brzucha, brak apetytu, zmniejszenie masy ciała) i inne. W szczególnie silnych przypadkach awitaminozy B1 może rozwinąć się choroba beri-beri, objawiająca się zaburzeniami pracy neuronów i włókien mięśniowych, powodującymi ból kończyn, osłabienie mięśni, drżenie, a ostatecznie niewydolność układu krążenia i nawet śmierć. Skutki nadmiaru tej substancji nie są dla nas szkodliwe. Tiamina posiada cechy katalityczne, co wykorzystamy w naszej pracy. Alkohol etylowy ma działanie szkodliwe na nasz organizm – pamiętajmy o tym mimo faktu, że jest to jedna z ulubionych trucizn człowieka już od wielu wieków. Wodorotlenek sodu jest silnie żrący. Należy zachować ostrożność jak zawsze przy pracy w laboratorium.
Przystępując do właściwej syntezy w kolbie umieszczamy 3,3g tiaminy, po czym rozpuszczamy ją w 9,5cm3 wody destylowanej. Witamina z łatwością rozpuszcza się w wodzie, a jej roztwór ma charakterystyczny, zdaniem wielu osób raczej przykry zapach. Po całkowitym rozpuszczeniu do roztworu dodajemy 25cm3 alkoholu etylowego, uzyskując klarowny i bezbarwny roztwór (Fot.2).

Przygotowany roztwór chłodzimy następnie w łaźni wodnej z kawałkami lodu i dodajemy po kropli, za każdym razem mieszając 9,5cm3 10% wodnego roztworu NaOH. Powoduje to wydzielenie się tiaminy w formie wolnej zasady, przez co roztwór przyjmuje żółte zabarwienie (Fot.3). Łaźnia zapobiega zbytniemu przegrzaniu roztworu, co mogłoby spowodować m.in. rozkład delikatnej witaminy.

Po ogrzaniu żółtawego roztworu do temperatury pokojowej zadajemy go, dodając po kropli 22cm3 aldehydu benzoesowego, ciągle mieszając. Rozpuszczalność tej substancji w wodzie jest znikoma, nawet dodatek alkoholu nie pozwala na całkowite rozpuszczenie. Z tego powodu po dodaniu całkowitej ilości aldehydu roztwór pozostaje mętny, ale jest to normalny objaw (Fot.4).

Po dokładnym wymieszaniu cieczy dalszą reakcję możemy prowadzić dwojako: w ciągu dwóch godzin w temperaturze 60°C (trzeba uważać, aby jej nie przekroczyć) lub w ciągu kilku dni w temperaturze pokojowej. Ja wypróbowałem obie metody i ich rezultat jest podobny. W opisywanym przypadku reakcja była prowadzona w temperaturze pokojowej w czasie czterech dni, bez ciągłego mieszania – kolbę pozostawiono zabezpieczoną przed światłem pod dygestorium. Wylot naczynia warto zabezpieczyć korkiem lub choćby przez owinięcie folią aluminiową, ponieważ połączony zapach witaminy i aldehydu dla niektórych osób może być wręcz obezwładniający.
Po wspomnianym czasie można było zaobserwować powstanie sporej ilości kryształów – jest to surowa benzoina C14H12O2 (Fot.5).

Mieszaninę należy schłodzić do około 0°C, uzyskaną benzoinę odsączyć, a następnie przemyć kilkukrotnie wodą destylowaną – pozwoli to usunąć pozostałości tiaminy, która jest dobrze rozpuszczalna w wodzie (w przeciwieństwie do benzoiny). Ostatnie płukanie należy przeprowadzić za pomocą 40-50% etanolu, aby usunąć ewentualne pozostałości nieprzereagowanego aldehydu benzoesowego. Produkt na sączku prezentuje się jako białe ciało stałe (Fot.6).

Do dalszych prac produkt warto jednak oczyścić przez krystalizację z etanolu – rozpuszczalność benzoiny w gorącym etanolu jest znacznie większa niż w niskiej temperaturze. Do rozpuszczenia na gorąco całej ilości otrzymanej benzoiny wystarczyło kilkadziesiąt centymetrów sześciennych etanolu 95%, a po ochłodzeniu wykrystalizowała już dużo czystsza benzoina (Fot.7).


Po odsączeniu i przemyciu zimnym 50% etanolem kryształy benzoiny wysuszono i zważono (Fot.8).

Przedstawiony proces to przykład kondensacji benzoinowej. Zwykle takie reakcje katalizuje się silnie toksycznymi cyjankami CN-, ale już w latach 50 ubiegłego wieku zaproponowano zastąpienie ich tiaminą, tak jak w opisanym przypadku (Rys.2).
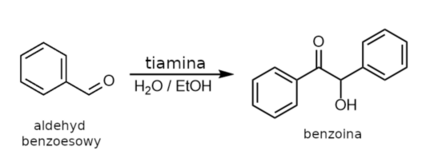
Jeśli chodzi o wydajność, to otrzymano 12,3g oczyszczonej benzoiny, co w przeliczeniu na wykorzystany aldehyd benzoesowy stanowi ~50% wydajności teoretycznej. Pozyskana benzoina została w większości wykorzystana na kolejnym etapie syntezy.
Jako cząstkową nagrodę za trudy warto wykorzystać niewielką część otrzymanej benzoiny w prostym doświadczeniu. Wystarczy do roztworu 0,2g benzoiny w 25cm3 alkoholu metylowego CH3OH (ostrożnie, trucizna) dodać 0,75g wodorotlenku potasu KOH w 1cm3 wody destylowanej. Początkowo bezbarwny roztwór (Fot.9A) już po chwili barwi się na jasnofioletowo (Fot.9B). Okazuje się jednak, że wstrząśnięcie roztworu powoduje zanik barwy, która pojawia się znowu po krótkim czasie, kiedy płyn pozostawimy w spokoju. Cykl ten można powtarzać wielokrotnie [9].

Zmiany barwy są powodowane utlenianiem benzoiny w środowisku alkalicznym przy udziale tlenu z powietrza.
Etap II – od benzoiny do benzilu
Tym razem potrzebujemy substancji z poniższej listy:
- benzoina C14H12O2,
- azotan amonu NH4NO3,
- octan miedzi(II) (CH3COO)2Cu,
- kwas octowy CH3COOH lodowaty (99%).
Benzoina to organiczny związek chemiczny z grupy aromatycznych ketoalkoholi. Jest stosowana m.in. w środkach zapachowych.
Azotan amonu, nazywany też saletrą amonową jest bezbarwnym, krystalicznym ciałem stałym. Jest higroskopijny i bardzo dobrze rozpuszcza się w wodzie. Azotan ten jest wykorzystywany jako składnik materiałów wybuchowych i przy produkcji nawozów mineralnych.
Przy pracy pamiętajmy, że stężony kwas octowy wykazuje dosyć silne działanie niszczące na nasze tkanki, a przy tym cechuje się drażniącym i duszącym zapachem. Nieco więcej uwagi musimy poświęcić octanowi miedzi(II). Substancję tę można oczywiście kupić w sklepie chemicznym, ale nie jest problematyczne samodzielne otrzymanie jej we własnej pracowni. W tym celu musimy przygotować nieco metalicznej miedzi, np. w formie ścinków nieizolowanego przewodu elektrycznego lub fragmentów blachy (Fot.10).

Skrawki miedzi zostały zalane około 100cm3 zwykłego spożywczego octu spirytusowego, będącego w istocie 6-10% roztworem kwasu octowego, po czym dodano do naczynia 50cm3 wodnego roztworu nadtlenku wodoru H2O2 o stężeniu 3% (aptecznej wody utlenionej). Przedstawione proporcje w żadnym razie nie są krytyczne i nawet duże odstępstwa od podanych ilości nie spowodują problemów z otrzymaniem produktu. Uzyskany roztwór jest początkowo bezbarwny (Fot.11). Układ reakcyjny należy zabezpieczyć przed parowaniem i pozostawić w temperaturze pokojowej na kilka dni lub delikatnie podgrzać, co znacznie przyspieszy reakcję.

Po pewnym czasie można zauważyć, że barwa roztworu się wyraźnie zmienia – reakcję możemy uznać za skończoną, kiedy płyn stanie się wyraźnie niebieski (Fot.12). Metaliczna miedź - jako użyta najpewniej w sporym nadmiarze - nie ulegnie całkowitemu roztworzeniu, można ją więc wykorzystać ponownie (przyp. aut.).

Po zlaniu płynu i odparowaniu części objętości wody (oraz pozostałości kwasu octowego) zaczynają z niego krystalizować piękne kryształki octanu miedzi(II) powstałe w myśl reakcji równania reakcji:
Uzyskany octan, po ewentualnym oczyszczeniu przez rekrystalizację oraz wysuszeniu może być wykorzystany w syntezie benzilu i prezentuje się jak na Fot.13.

Wracając jednak do syntezy benzilu, do kolby okrągłodennej musimy wprowadzić 11g otrzymanej uprzednio benzoiny, 31,4cm3 lodowatego kwasu octowego, 5g azotanu amonu i 6,3cm3 roztworu octanu miedzi(II) przygotowanego przez rozpuszczenie 1,1g tej substancji w 50cm3 wody destylowanej. Kolbę z mętną na tym etapie mieszaniną (w temperaturze pokojowej nie wszystkie reagenty ulegają rozpuszczeniu) umieszczamy następnie w płaszczu grzewczym i instalujemy chłodnicę zwrotną, co można zobaczyć na Fot.14.

Grzanie i mieszanie należy ustawić tak, by mieszanina wrzała delikatnie przez czas około 1,5 godziny. W tym czasie reagenty stopniowo rozpuszczają się, a mieszanina reakcyjna przyjmuje zieloną barwę (Fot.15).


Po upływie założonego czasu należy pozwolić mieszaninie ostygnąć, najlepiej do następnego dnia. W tym czasie ulegnie ona częściowemu zestaleniu na skutek krystalizacji (Fot.16).

Uzyskany benzil C14H10O2 należy odsączyć, po czym przynajmniej kilkukrotnie przemyć na sączku wodą destylowaną, a na koniec niewielką porcją zimnego etanolu.
Prezentowana reakcja zachodzi według schematu jak na Rys.3 [10].

Po wysuszeniu i sprawdzeniu zgodności temperatury topnienia produktu z wartością przytaczaną w literaturze (94–95°C) uznałem, że nie jest konieczne dalsze jego oczyszczanie przez rekrystalizację, co prowadziłoby do zwiększenia strat [11]. Benzil w warunkach normalnych to żółte ciało stałe (Fot.17).

Otrzymano 10,45g benzilu o jakości wystarczającej do dalszych prac, co w przeliczeniu na wykorzystaną benzoinę stanowi 96% wydajności teoretycznej.
Etap III – od bezilu do lofiny
Przed nami ostatni krok na drodze do syntezy tak pożądanej przez nas substancji. W tym przypadku musimy przygotować:
- benzil C14H10O2,
- aldehyd benzoesowy C6H5CHO,
- octan amonu CH3COONH4,
- kwas octowy CH3COOH lodowaty (99%).
Benzil, nazywany też dibenzoilem to aromatyczny diketon, który można rozpatrywać jako dimer grupy benzoilowej. Używany jest jako produkt pośredni w syntezie organicznej oraz jako fotoinicjator wolnorodnikowego utwardzania polimerów. Może mieć negatywne działanie na organizmy żywe. Octan amonu jest natomiast solą amonową kwasu octowego i bywa wykorzystywany w przemyśle spożywczym jako dodatek E264, czyli jeden z konserwantów.
Technicznie rzecz biorąc ten etap syntezy jest podobny do poprzedniego [12] [13]. W kolbie okrągłodennej umieszczamy 10,4g benzilu, 5,2cm3 aldehydu, 46,2g octanu amonu oraz 144cm3 lodowatego kwasu octowego. Zestaw umieszczamy w płaszczu grzewczym i prowadzimy ogrzewanie z mieszaniem w temperaturze delikatnego wrzenia przez czas trzech godzin. Roztwór początkowo jest wyraźnie żółty (Fot.18).


Wraz z postępem reakcji barwa cieczy staje się coraz mniej intensywna (Fot.19).

Po wyłączeniu ogrzewania musimy pozostawić mieszaninę do wystygnięcia, a następnie wlać cienkim strumieniem do przynajmniej litra intensywnie mieszanej zimnej wody destylowanej. Podczas dodawania mieszaniny do wody zaczynają się natychmiast wytrącać duże ilości białego osadu – jest to surowa lofina C21H16N2 (Fot.20).


Surową lofinę po odsączeniu należy kilkukrotnie przemyć wodą, a potem wysuszyć. Otrzymałem 13,6g surowej lofiny w postaci białego puszystego proszku (Fot.21).

Lofinę warto oczyścić przez rekrystalizację z gorącego metanolu – można także zastosować etanol lub izopropanol, ale potrzebne są ich większe ilości. Pięknie prezentują się igiełkowate kryształki lofiny powstające m.in. na ściankach naczynia w alkoholowym roztworze podczas jego powolnego stygnięcia (Fot.22).

Oczyszczona lofina po odsączeniu została wysuszona i przechowana do dalszych doświadczeń (Fot.23). Zebrany przesącz zawiera w dalszym ciągu nieco lofiny – warto go więc wykorzystać do pierwszych prób z chemiluminescencją, o czym napiszę nieco dalej.

Reakcja syntezy lofiny przebiega według schematu ukazanego na Rys.4.
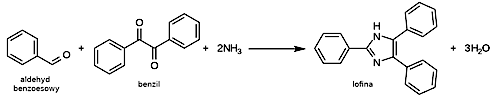
Masa lofiny po rekrystalizacji została wyznaczona jako 10,6g, co w przeliczeniu na wykorzystany benzil stanowi 72,3% wydajności teoretycznej.
Finał – chemiluminescencja
W celu obserwacji chemiluminescencji lofiny potrzebujemy:
- wodorotlenek sodu NaOH lub potasu KOH,
- nadtlenek wodoru H2O2 3% (apteczna woda utleniona),
- chloran(I) sodu NaClO.
Chloran(I) sodu jest – szczęśliwie dla eksperymentatora - łatwy do zdobycia i nie trzeba go wcale kupować w specjalistycznym sklepie chemicznym. Związek ten jest składnikiem wielu wybielaczy (nazywanych chlorowymi) stosowanych w gospodarstwie domowym, szczególnie tych najtańszych.
Tym razem chciałbym zaproponować uproszczoną procedurę przygotowania reakcji chemiluminescencji lofiny. W tym celu w 50cm3 metanolu (lub etanolu) należy rozpuścić 0,1g lofiny, po czym dodać bardzo niewielką ilość wodorotlenku sodu lub potasu – wystarczy kilka granulek wielkości główki szpilki. Wodorotlenek nie rozpuści się całkowicie, ale zalkalizuje roztwór w odpowiednim stopniu. Następnie trzeba dodać 10cm3 nadtlenku wodoru w postaci aptecznej wody utlenionej. Roztwór taki jest nietrwały i najlepiej przygotowywać go bezpośrednio przed wykorzystaniem. Osobno przygotowujemy 40cm3 roztworu wybielacza zawierąjacego chloran(I) przez jego rozcieńczenie równą objętością wody destylowanej.
W celu prezentacji najlepiej zaciemnić pomieszczenie, po czym wlewać wolnym strumieniem roztwór chloranu(I) do alkoholowego roztworu lofiny z dodatkiem wodorotlenku sodu i nadtlenku wodoru (Fot.24A). Przydatne jest mieszadło magnetyczne

Podczas reakcji możemy zobaczyć emisję jasnego żółtozielonego światła (Fot.24B). Przesącz zachowany po rekrystalizacji lofiny warto wykorzystać także do demonstracji chemiluminescencji w ten sam sposób (Fot.25). Na końcu strony, w dziale uzupełniej autorskich umieściłem film prezentujący chemiluminescencję pozostałości lofiny w przesączu (przyp. aut.).

Wyjaśnienie
Warto zaznaczyć, że lofina jest jedną z pierwszych otrzymanych sztucznie substancji chemicznych wykazujących efekt chemiluminescencji. Lofina jako 2,4,5-trifenylimidazol należy do grupy związków chemicznych nazywanych imidazolami. W środowisku alkalicznym, w obecności jonów podchlorynowych ClO- lofina jest utleniana przez nadtlenek wodoru. Jeden z atomów azotu posiada luźno związany atom wodoru, o właściwościach kwasowych. Drugi atom azotu z kolei ma właściwości zasadowe, więc lofina jako całość jest związkiem amfoterycznym. Podczas utleniania dochodzi do powstania mostka nadtlenowego; utworzony cykliczny nadtlenek jest bardzo nietrwały. Szybko dochodzi do jego rozpadu. Zgodnie z zasadą zachowania nadwyżka energii zostaje oddana do środowiska, tutaj pod postacią światła o długości fali λ=525nm. Światło takie ma barwę zielonkawo-żółtą.
Wiemy, że wydajność całkowitą procesu wieloetapowego można wyznaczyć jako iloczyn wydajności cząstkowych. W ten sposób możemy powiedzieć, że wydajność naszej syntezy, zaczynając od aldehydu benzoesowego wyniosła około 35%, na co największy wpływ miał proces o najniższej wydajności własnej (etap I, efekt wąskiego gardła). Tak czy inaczej jest to wydajność wielokrotnie wyższa (w tym przypadku 10-20 razy) niż w przypadku metody historycznej, gdzie lofina była otrzymywana na drodze cyklizacji hydrobenzamidu przy pomocy tlenu atmosferycznego. Myślę więc, że mimo poniesionych trudów - które też mają przecież aspekt dydaktyczny - nasze starania będą opłacalne.
Chemiluminescencja lofiny ma pewne znaczenie w analityce chemicznej i nie jest jedynie ciekawostką.
Literatura:
- [1] Ples M., Błękitna poświata. Synteza i chemiluminescencja związku Grignarda, Chemia w Szkole, 6 (2017), Agencja AS Józef Szewczyk, str. 14-17 powrót
- [2] Ples M., Całkiem niezwykła herbatka, Chemia w Szkole, 4 (2015), Agencja AS Józef Szewczyk, str. 6-9 powrót
- [3 Ples M., Fiolet świeci - chemiluminescencja powszechnie dostępnego związku manganu, Chemia w Szkole, 6 (2018), Agencja AS Józef Szewczyk, 16-19 powrót
- [4] Ples M., Światło z retorty, Chemia w Szkole, 5 (2014), Agencja AS Józef Szewczyk, str. 33-34 powrót
- [5] Ples M., Chemiluminescencja metalicznego sodu, Chemia w Szkole, 1 (2014), Wydawnictwo EduPress, str. 5-7 powrót
- [6] Ples M., Synteza i chemiluminescencja lofiny - zimne światło, muzyka i migdały, Chemia w Szkole, 5 (2020), Agencja AS Józef Szewczyk, str. 44-47 powrót
- [7] Dzieleńdziak A., Benzoina (kondensacja benzoinowa katalizowana tiaminą - witaminą B1), instrukcja do ćwiczeń, dostępne online: https://www.chem.umk.pl/panel/wp-content/uploads/Benzoina.pdf [dostęp 25.11.2022] powrót
- [8] Thiamin. Fact Sheet for Consumers, w: Dietary Supplement Fact Sheets, National Institutes of Health, 2015, dostępne online: https://ods.od.nih.gov/factsheets/Thiamin-Consumer/ [dostęp 25.11.2022] powrót
- [9] Pluciński T., Doświadczenia chemiczne, Wydawnictwo Adamantan, 1997, str. 69 powrót
- [10] Depreux P., Bethegnies G., Marcincal-Lefebvre A., Synthesis of benzil from benzoin with copper(II) acetate, Journal of Chemical Education, 1988, 65(6), str. 553 powrót
- [11] Farmakopea Polska X, Polskie Towarzystwo Farmaceutyczne, Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, Warszawa, 2014, s. 4276 powrót
- [12] Mickiewicz D., Lofina – luminofor prosty do otrzymania w domu, dostępne online: https://biomist.pl/chemia/doswiadczenia-chemiczne/lofina-luminofor-prosty-do-otrzymania-w-domu/2426 [dostęp 25.11.2022] powrót
- [13] Radziszewski B. R., Untersuchungen über Hydrobenzamid, Amarin und Lophin, Berichte der deutschen chemischen Gesellschaft, 10 (1), 1877, str. 70–75 powrót
Wszystkie fotografie i rysunki zostały wykonane przez autora
Uzupełnienie autora
Chemiluminescencję pozostałości lofiny w przesączu pozostałym po jej rekrystalizacji możemy zobaczyć na poniższym filmie:
Marek Ples