Pomocna dłoń chemii w rękawiczce - synteza luminolu z odpadów
Poniższy artykuł został opublikowany pierwotnie w czasopiśmie dla nauczycieli Chemia w Szkole (5/2023):

Chemia, jako nauka o strukturze, składzie oraz przemianach materii, otwiera przed nami fascynujący świat mikro- i makrokosmosu. To dziedzina, która w sposób niezwykły wpływa na nasze życie codzienne, ponieważ zakres jej zainteresowań jest szczególnie szeroki: od procesów zachodzących w organizmach, aż po takie, które kształtują cały otaczający nas świat. Edukacja w dziedzinie chemii nabiera szczególnego znaczenia poprzez praktyczne eksperymenty, które pozwalają uczniom na samodzielne doświadczanie zjawisk chemicznych. Dlaczego eksperymenty w nauczaniu są tak kluczowe? Odpowiedź tkwi w naturze samej chemii, która bywa abstrakcyjna i trudna do zrozumienia jedynie na podstawie teorii. Eksperymenty pozwalają uczniom doświadczyć za pomocą własnych zmysłów, jak reakcje chemiczne zachodzą w praktyce, jakie zmiany obserwujemy oraz jakie są ich konsekwencje. To właśnie przez eksperymenty uczniowie mogą zetknąć się z magią chemii i zobaczyć, jakie potężne narzędzie naukowe tkwi w ich rękach.
Jednym z najbardziej pociągających aspektów eksperymentów chemicznych są reakcje chemiluminescencyjne. Te wyjątkowe procesy wydzielają światło w wyniku reakcji chemicznych, tworząc widowiskowe efekty w ciemności. Dla nauczycieli stanowią one niezwykłe narzędzie dydaktyczne, umożliwiające zaintrygowanie uczniów oraz rozbudzenie ich ciekawości. Dla uczniów natomiast, reakcje chemiluminescencyjne to nie tylko fascynujące doświadczenie, ale także doskonała okazja do praktycznego zetknięcia się z abstrakcyjnymi koncepcjami chemicznymi, takimi jak przemiany energii na poziomie molekularnym.
Istnieje wiele substancji o właściwościach chemiluminescencyjnych, takich jak biała odmiana alotropowa fosforu P4, lucygenina C28H22N4O6, lofina C21H16N2, związki magnezo- i krzemoorganiczne oraz wiele innych [1] [2] [3] [4]. Jedną z najbardziej znanych substancji tego typu jest luminol, który jednak nie należy do szczególnie powszechnie spotykanych związków chemicznych [5]. Okazuje się jednak, że tę ciekawą substancję można wytworzyć stosując jako jeden z surowców… zużyte rękawiczki jednorazowe, co opiszę dalej. Opisana synteza jest etapowa, a jej zwieńczeniem będzie wytworzenie wspomnianego związku chemiluminescencyjnego.
Zapraszam Szanownego Czytelnika do powtórzenia opisanych doświadczeń. Chcę jednak stanowczo zaznaczyć, że jest przy tym konieczna zdrowa doza ostrożności, ponieważ będziemy pracować z substancjami w różny sposób niebezpiecznymi, zarówno żrącymi i drażniącymi (kwasy, zasady, nadtlenek wodoru), jak i toksycznymi oraz gwałtownie reagującymi. Konieczne są normalnie środki ochrony osobistej. Radzę także przestrzegać wszystkich podanych w tekście wskazówek.
Etap I – Otrzymywanie bezwodnika ftalowego
Rozpoczynając naszą pracę musimy zgromadzić substancje takie jak:
- rękawiczki jednorazowe winylowe – około 500g (100 par)
- wodorotlenek sodu – 100g
- kwas chlorowodorowy stężony HCl(aq)– 250cm3
Jednorazowe rękawiczki winylowe (Fot.1) traktujemy w tym przypadku jako odczynnik, surowiec wyjściowy do produkcji pożądanej przez nas substancji.

Co prawda, potrzebna ilość jest dosyć spora – z powodu niewielkiej zawartości poszukiwanego substratu – ale rękawiczki są stosunkowo tanie. Dodatkowo, nie muszą być to rękawiczki nowe, wprost z opakowania. Opisaną tu procedurę przeprowadziłem z wykorzystaniem rękawiczek zużytych podczas normalnych prac w laboratorium, zbieranych przez dłuższy czas w przeznaczonym do tego pojemniku. Oczywiście, należy przy tym pamiętać, aby wykorzystane rękawiczki nie były zanieczyszczone silnie toksycznymi chemikaliami. Myślę też, że tego typu ponowne wykorzystanie odpadów ma walor edukacyjny w zakresie ekologii. W każdym razie, rękawiczki wypłukano dwukrotnie w wodzie i wysuszono, po czym pocięto na niewielkie fragmenty.

Potrzebna do przetworzenia ilość surowca jest na tyle duża, że warto podzielić ją na mniejsze porcje – ja jednorazowo wykorzystałem około 167g pociętych rękawiczek, wrzucając je do kolby okrągłodennej i zalewając około 800cm3 alkoholu izopropylowego C3H7OH po czym ogrzewając do wrzenia pod chłodnicą zwrotną w ciągu 2 godzin (Fot.3). Cały proces powtórzyłem więc trzykrotnie.
Zdaję sobie sprawę, że ilość potrzebnego alkoholu jest dosyć spora, ale po przetworzeniu każdej porcji surowca, rozpuszczalnik może zostać oddestylowany z mieszaniny poreakcyjnej i wykorzystany ponownie.

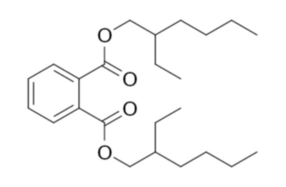
Tworzywo sztuczne, z którego wykonane są rękawiczki winylowe samo w sobie jest twarde i kruche. Aby nadać mu elastyczność stosuje się dodatki różnorodnych specyficznych substancji, czyli tak zwanych plastyfikatorów. Korzystnie dla nas jedną z częściej wykorzystywanych substancji tego typu jest ester kwasu ftalowego, a dokładniej ftalan di(2-etyloheksylu) C24H38O4 (Rys.1) [6]. Zaznaczę jednak, że zdarzają się także inne plastyfikatory, w tym bez udziału ftalanów – wtedy należy poszukać innego surowca.
Po upływie wspomnianego czasu należy oziębić mieszaninę i zlać płyn zawierający między innymi plastyfikator, odrzucając stałe pozostałości tworzywa sztucznego. Płyn zadajemy następnie około 33g wodorotlenku sodu rozpuszczonymi w 800cm3 wody destylowanej (w tym czasie często jest zauważalne wyraźne zmętnienie mieszaniny, vide Fot.4), po czym ogrzewamy ponownie do wrzenia pod chłodnicą zwrotną w czasie jednej godziny.

Na tym etapie ma miejsce hydroliza estru w środowisku zasadowym. Po ochłodzeniu i pozostawieniu mieszaniny w spokoju dochodzi szybko do wyraźnego rozdzielenia faz (Fot.5). Dolna faza wodna zawiera potrzebną substancję, zaś górna alkoholowa pozostałe produkty reakcji (w tym izopropanol, który możemy odzyskać do ponownego wykorzystania).

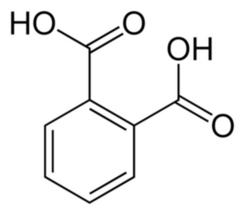
Dolną fazę odzyskujemy w rozdzielaczu i następnie zadajemy 84cm3 stężonego kwasu chlorowodorowego, po czym schładzamy w lodówce. Działanie tego mocnego kwasu powoduje wytrącenie białych kryształków wolnego kwasu ftalowego C8H6O4(Fot.6, Rys.2) z jego soli.

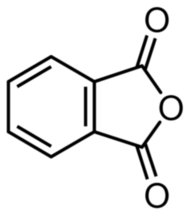
Niestety, tak uzyskany kwas ftalowy jest dosyć silnie zanieczyszczony. Możemy go jednak łatwo oczyścić. Wystarczy odsączyć i wysuszyć uzyskaną substancję, a następnie przenieść ją na dno dużej, wysokiej zlewki, którą nakrywamy kolbą okrągłodenna z zimną wodą. Po ogrzaniu kwas ftalowy przekształca się do swojego bezwodnika C8H4O3 (Rys.3).
W podwyższonej temperaturze bezwodnik ftalowy sublimuje, po czym zestala się w kontakcie z zimną powierzchnią kolby. W ten sposób możemy zebrać już bardzo czyste, olśniewająco białe kryształy tego związku w formie delikatnych igiełek (Fot.7).

Uzyskałem 31,2g oczyszczonego bezwodnika ftalowego, który został wykorzystany do dalszej syntezy. Trudno w tym przypadku mówić o rzeczywistej wydajności procesu, ponieważ problematyczne jest uzyskanie informacji o początkowej zawartości plastyfikatora w rękawiczkach. Możemy stwierdzić jednak, że uzyskana substancja stanowi około 6,2% masowych surowca wyjściowego. Jest to oczywiście niewiele, ale biorąc pod uwagę fakt, że związek ten otrzymaliśmy z odpadów, używając do tego dodatkowo tanich i łatwodostępnych substancji pomocniczych, gra wydaje się być warta świeczki.
Etap II – Od bezwodnika ftalowego do kwasu 3-nitroftalowego
W tym etapie syntezy naszym celem jest wprowadzenie grupy nitrowej −NO2 do cząsteczki bezwodnika ftalowego, a także uzyskanie odpowiedniego kwasu. W tym celu w kolbie stożkowej umieśćmy 30g suchego bezwodnika ftalowego, po czym – ciągle mieszając, aby uniknąć zbrylenia – dodajmy 28,5cm3 stężonego kwasu azotowego(V) HNO3. Po dokładnym rozprowadzeniu substancji stałej w cieczy rozpoczynamy powolne wprowadzanie do mieszaniny 28,5cm3 stężonego kwasu siarkowego(VI) H2SO4 – proces ten jest egzotermiczny, trzeba więc zwracać uwagę, by nie doszło do niekontrolowanego wrzenia. Podczas wkraplania kwasu konieczne jest intensywne mieszanie, ale musimy uważać, aby nie rozpryskiwać żrącego płynu. Początkowo trudno zauważyć jakiekolwiek oznaki zachodzenia reakcji, ponieważ uzyskujemy po prostu zawiesinę białej substancji krystalicznej w mieszaninie kwasów (Fot.8).

Przygotowaną mieszaninę wstawiamy do łaźni wodnej o temperaturze bliskiej 100°C. Szczęśliwie dla nas, w tym przypadku reakcja nitrowania przebiega zwykle raczej spokojnie, co jednak nie oznacza, że możemy zaniechać środków ostrożności. Już po chwili ogrzewania staną się widoczne brunatne tlenki azotu wydzielające się z mieszaniny (Fot.9). Są one silnie toksyczne, więc reakcja musi być prowadzona pod BARDZO wydajnie działającym wyciągiem lub na zewnątrz.

sita molekularne w łaźni wodnej jako ewentualne zarodki wrzenia
Ogrzewanie musimy prowadzić w czasie 2 godzin, po czym ostudzić mieszaninę i wlać ją powoli (proces egzotermiczny, ciągle mieszać!) do około 70cm3 wody destylowanej. Na dnie naczynia zbierze się osad produktu (Fot.10).

Myślę, że warto tutaj nadmienić, że w czasie nitrowania bezwodnika kwasu ftalowego w podanych warunkach dochodzi do powstania dwóch izomerów kwasu nitroftalowego (odpowiednie bezwodniki przyłączają w międzyczasie wodę), a mianowicie 3-nitroftalowego (Rys.4A) i 4-nitroftalowego (Rys.4B).
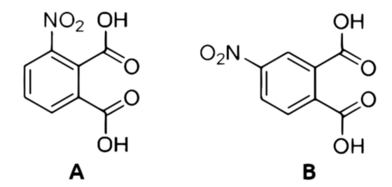
Substancje te dosyć łatwo rozdzielić, ponieważ różnią się wyraźnie rozpuszczalnością: izomer z grupą nitrową przyłączoną w pozycji trzeciej jest dużo słabiej rozpuszczalny niż izomer 4-nitro, a więc to właśnie ten pierwszy wydzielił się z roztworu w postaci osadu. Osad ten wystarczy następnie kilkukrotnie przemyć zimną wodą, a potem przekrystalizować z niej na gorąco i wysuszyć. Otrzymuje się w ten sposób praktycznie czysty kwas 3-nitroftalowy w postaci białego ciała krystalicznego (Fot.11)

Uzyskałem 13,2g pożądanego kwasu, co stanowi wydajność około 31% w przeliczeniu na wykorzystany bezwodnik ftalowy. Wydajność nie jest wysoka, ale wytłumaczeniem tego jest fakt, że drugim – niewykorzystanym przez nas i nie ujętym w tych obliczeniach – produktem jest kwas 4-nitroftalowy.
Etap IIa – Synteza siarczanu hydrazyny
Aby przekształcić kwas ftalowy w jego hydrazyd, musimy dokonać jego kondensacji z hydrazyną N2H4. Substancja ta, nazywana też diazanem, to nieorganiczny związek chemiczny zbudowany z dwóch połączonych ze sobą wiązaniem N–N grup aminowych. Jest to bardzo silnie trująca, bezbarwna, łatwopalna, żrąca i higroskopijna ciecz, dymiąca na powietrzu. Cząsteczka hydrazyny ma duży moment dipolowy, co świadczy o tym, że występuje ona głównie w konformacji gauche, w której obie grupy aminowe są skręcone o kąt ok. 90–95° w stosunku do symetrycznej konformacji naprzeciwległej [7] [8].
Toksyczność hydrazyny nie zachęca do pracy z tą substancją i do prowadzenia jej syntezy. Jest jednak inny sposób: zamiast stosować wolną hydrazynę, użyjemy jej soli: siarczanu(VI), czy raczej wodorosiarczanu(VI) hydrazyny N2H6SO4. W temperaturze normalnej jest to białe ciało krystaliczne, którym dużo łatwiej i bezpieczniej manipulować niż czystą hydrazyną.
Aby dokonać syntezy wodorosiarczanu(VI) hydrazyny potrzebujemy:
- chloran(I) sodu (podchloryn sodu) NaClO, roztwór wodny 5% – 500cm3
- wodorotlenek sodu NaOH – 32g
- mocznik CH4N2O – 22g
- żelatyna – 0,75g
- kwas siarkowy(VI), 50% – 100cm3
Chloran(I) sodu to substancja dostępna dla eksperymentatora i nie ma konieczności jej zakupu w specjalistycznym sklepie chemicznym. Związek ten jest składnikiem wielu środków wybielających (określanych jako chlorowe), które znajdują zastosowanie w domowych gospodarstwach. Do doświadczenia możemy zastosować praktycznie każdy chlorowy wybielacz o koncentracji chloranu zbliżonej do podanej – najlepsze są te najtańsze, ponieważ nie zawierają one dużej ilości innnych dodatków.
Mocznik, znany także jako karbamid, to związek organiczny o fundamentalnym znaczeniu w biologii i chemii. Jest produktem przemiany materii organizmów, zwłaszcza w wyniku metabolizmu białek. Mocznik jest także powszechnie stosowany w przemyśle chemicznym, rolniczym oraz farmaceutycznym. W rolnictwie, jako nawóz azotowy, ma kluczowe znaczenie dla wzrostu i wydajności roślin. W przemyśle farmaceutycznym wykorzystywany jest m.in. w produkcji leków oraz kosmetyków. Ponadto, w laboratoriach medycznych pełni funkcję wskaźnika funkcjonowania nerek, co sprawia, że jest niezmiernie istotnym elementem w diagnostyce medycznej. W warunkach normalnych ma postać białych kryształów.
Przystępując do syntezy musimy schłodzić w dużym naczyniu 500cm3 roztworu podchlorynu. Najlepsza w tym celu jest kąpiel lodowa, ponieważ na pierwszym etapie syntezy konieczne jest utrzymywanie temperatury zbliżonej do 8°C (Fot.12).

Ważne jest aby pojemność naczynia była dużo większa niż cieczy, ponieważ w czasie reakcji może dochodzić do silnego pienienia się.
Następnie do ciągle mieszanego roztworu dodajemy powoli, małymi porcjami 32g wodorotlenku sodu, ciągle utrzymując temperaturę na zadanym poziomie – w żadnym momencie nie powinna ona wyraźnie przekroczyć 10-15°C, ponieważ spowoduje to gwałtowne przyspieszenie rozkładu podchlorynu.
Osobno przygotowujemy roztwory:
- 0,75g żelatyny w 15-18cm3 wody destylowanej
- 22g mocznika w 40cm3 wody destylowanej
W obu przypadkach dokładne rozpuszczenie substancji może wymagać ogrzania roztworów, które następnie łączymy i delikatnie chłodzimy.
Usuwamy kąpiel lodową i do ochłodzonego do 8°C (ta temperatura jest odpowiednia, nie jest celowe silniejsze chłodzenie) wlewamy naraz całą objętość roztworu mocznikowo-żelatynowego, po czym naczynie przykrywamy – konieczne jest jednak ciągłe i bardzo silne mieszanie, więc mieszadło magnetyczne jest w tym przypadku wręcz idealnym rozwiązaniem. Roztwór zmienia w tym czasie barwę na żółtawą oraz mętnieje – powstają przy tym duże ilości ulatniającego się gazu.
W tym czasie w układzie reakcyjnym dochodzi do bardzo interesującego procesu, jakim jest przegrupowanie Hofmanna. Jest to reakcja chemiczna, w której pierwszorzędowe amidy ulegają przekształceniu do pierwszorzędowych amin o łańcuchu węglowym krótszym o jeden atom węgla, przy czym zostaje uwolniony także dwutlenek węgla CO2 [9] [10]. Bardzo ważne jest przykrycie naczynia i prowadzenie reakcji pod wyciągiem lub na zewnątrz, ponieważ dochodzi do uwolnienia niewielkich ilości par hydrazyny.
Warto wspomnieć kilka słów o roli żelatyny, ponieważ jest ona tutaj kluczowa. Przegrupowanie Hofmanna jest procesem bardzo czułym na zanieczyszczenie środowiska reakcji jonami metali. Aby umożliwić reakcję muszą one zostać spułapkowane, do czego wykorzystuje się związki kompleksujące takie jak kwas etylenodiaminotetraoctowy C10H16N2O8 (wersenowy) i jego sole. Podobne właściwości ma jednak żelatyna, która okazała się całkowicie wystarczająca w tym zastosowaniu.
Roztwór mieszamy aż do momentu, kiedy tempo ewakuacji gazu wyraźnie się zmniejszy, a potem ogrzewamy do 85°C w czasie pięciu minut. Z powodu obniżenia rozpuszczalności gazu w podwyższonej temperaturze z roztworu uchodzi pozostały dwutlenek węgla (Fot.13).

W dalszej kolejności ciecz pozostawiamy do wystygnięcia i chłodzimy do temperatury 0°C, by potem dodać powoli, kroplami (na początku bardzo delikatnie, potem nieco szybciej) całą objętość kwasu siarkowego(VI). Ostrożność jest konieczna, ponieważ ma miejsce wtedy uwalnianie dużych ilości gazu oraz silne pienienie. Zakwaszenie środowiska początkowo powoduje zobojętnienie wodorotlenku oraz węglanu sodu, a następnie przekształcenie wolnej hydrazyny w jej sól, która po ochłodzeniu wypada z roztworu w postaci białych kryształków (Fot.14).

Kryształy odsączamy, przemywamy małą ilością lodowatej wody i pozostawiamy do wyschnięcia w temperaturze pokojowej. Produkt jest zanieczyszczony niewielką ilością siarczanu(VI) sodu Na2SO4, ale nie istnieje prosta metoda dalszego oczyszczenia soli hydrazyny z powodu podobnej rozpuszczalności obu substancji w najbardziej popularnych rozpuszczalnikach. Nie przeszkodzi nam to jednak w dalszej syntezie.
Otrzymałem 23g soli, co stanowi 47,7% w przeliczeniu na wykorzystane substraty. Otrzymany produkt należy przechowywać w suchych warunkach, w ciemności.
Etap IIb – Synteza octanu sodu
Następną substancją jakiej potrzebujemy na dalszych etapach syntezy jest octan sodu. Jest to sól kwasu octowego i sodu o wzorze chemicznym CH3COONa. Jest powszechnie wykorzystywany zarówno w przemyśle, jak i gospodarstwach domowych. Jego właściwości sprawiają, że jest używany jako środek konserwujący oraz regulator pH. W branży spożywczej octan sodu pełni rolę konserwantu, który pomaga utrzymać świeżość i trwałość różnych produktów, takich jak marynaty, konserwy czy przetwory warzywne. Ponadto, jest również stosowany w przemyśle tekstylnym, skórzanym i papierniczym. Możemy więc powiedzieć, że octan ten jest związkiem o wszechstronnym zastosowaniu, odgrywając istotną rolę zarówno w produkcji przemysłowej, jak i w codziennym życiu.
Octan sodu jest substancją stosunkowo łatwodostępną i tanią, ale można go też otrzymać wykorzystując dwie substancje dostępne praktycznie w każdym domu lub możliwe do zakupienia w sklepie spożywczym. Są to:
- ocet spirytusowy (kwas octowy CH3COOH, 6-10%)
- soda oczyszczona (wodorowęglan sodu NaHCO3)
Zamiast octu, możemy wykorzystać tzw. esencję octową, która jest roztworem kwasu octowego o stężeniu 70-80%, co jest nawet korzystniejsze.
W dużym naczyniu (ryzyko pienienia) umieszczamy 0,5dm3 octu spirytusowego, a następnie dodajemy do niego sodu oczyszczonej, póki powoduje to uwalnianie dużych ilości gazowego dwutlenku węgla. Należy unikać zbyt dużego dodatku wodorowęglanu. W czasie zobojętniania kwasu octowego dochodzi do powstania potrzebnego nam octanu sodu. By go uzyskać, roztwór odparowujemy tak długo, aż zmętnieje i zaczną się wydzielać białe kryształy (Fot.15).

Teraz w dalszym ciągu ogrzewając dodajemy do naczynia tyle wody, aby po wymieszaniu rozpuścić wydzielone kryształy. W ten sposób uzyskujemy klarowny roztwór, który szybko przesączamy na gorąco do czystego naczynia, nakrywamy i pozostawiamy do wystygnięcia.
Przy okazji dalszych prac warto zaobserwować ciekawe właściwości octanu sodu. Okazuje się, że możemy ochłodzić klarowny, nasycony roztwór tej substancji, przy czym octan nie ulegnie zestaleniu, chociaż jest on w temperaturze pokojowej ciałem stałym. Powstaje metastabilna ciecz przechłodzona, którą możemy następnie wytrącić z tego stanu np. poprzez wstrząs lub wprowadzenie zarodka krystalizacji w postaci niewielkiego kryształku stałej substancji (Fot.16A).

A – zarodnikowanie niewielkim kryształkiem octanu (0s),
B – początek krystalizacji (2s),
C – cała objętość roztworu skrystalizowana (5s)
Zaobserwujemy w ten sposób bardzo szybki proces krystalizacji trójwodnego octanu sodu: strefa krystalizacji rozprzestrzenia się wręcz w oczach (Fot.16B) i w ciągu kilku sekund cała objętość cieczy przekształca się w ciało stałe (Fot.16C). Proces ten jest bardzo widowiskowy. Jednocześnie do otoczenia zostaje oddane ciepło przemiany fazowej, co bywa wykorzystywane w wielorazowych ogrzewaczach dłoni.
Tak uzyskaną bryłę możemy pokruszyć, kryształy podsuszyć i wykorzystać do dalszych doświadczeń (Fot.17). Wydajność reakcji w tym wypadku przekracza 90%.

Etap III – Od kwasu 3-nitroftalowego do hydrazydu 3-nitroftalowego
Na tym etapie przydadzą się nam głównie zsyntezowane uprzednio substancje:
- kwas 3-nitroftalowy – 13g
- wodorosiarczan(VI) hydrazyny – 13g
- trójwodny octan sodu – 13g
- gliceryna (glicerol) – 65cm3
Gliceryna C3H8O3 to organiczny związek chemiczny z grupy cukroli; najprostszy trwały alkohol trójwodorotlenowy (triol). W warunkach normalnych jest syropowatą bezbarwną cieczą, silnie higroskopijną. Zastosowanie gliceryny jest bardzo szerokie, od syntezy chemicznej, przez wykorzystanie jako rozpuszczalnik, aż do przemysłu spożywczego.
W szklanym naczyniu musimy umieścić wszystkie substancje stałe, po czym zalać je 52cm3 wody destylowanej. Chemikalia w temperaturze pokojowej nie ulegają w tym wypadku całkowitemu rozpuszczeniu, więc zostaje uformowana biała zawiesina (Fot.18).

Mieszaninę następnie ogrzewamy delikatnie, ciągle mieszając, aż substancje stałe ulegną rozpuszczeniu, na skutek czego powstanie żółtawy klarowny płyn. Wtedy do roztworu wlewamy wskazaną objętość gliceryny (o jak najmniejszej zawartości wody). Lepki żółtawy roztwór (Fot.19) musimy dobrze wymieszać i rozpocząć jego powolne ogrzewanie, przy ciągłym kontrolowaniu temperatury.

Po co w roztworze gliceryna? Okazuje się, że reakcja kondensacji hydrazyny (wyzwolonej in situ ze swojej soli) z kwasem 3-nitroftalowym zachodzi wydajnie dopiero w temperaturach znacząco wyższych od możliwych do uzyskania w roztworze wodnym przy normalnym ciśnieniu. Gliceryna pełni tu więc rolę wysokowrzącego środowiska reakcji. Aby było to jednak możliwe, musimy z układu usunąć prawie całą wodę. Tak więc kontynuując ogrzewanie możemy zauważyć, że w temperaturach z zakresu 100-120°C dochodzi do silnego pienienia się mieszaniny spowodowanego uchodzeniem pary wodnej. Po usunięciu całej wody temperatura zacznie szybko wzrastać – naszym celem jest utrzymanie jej w granicach 200-220°C w czasie pięciu minut. Musimy przy tym unikać przegrzania, ponieważ może to spowodować rozkład produktu. W tym czasie dochodzi do wytrącenia dużych ilości brudnożółtego lub nawet pomarańczowego produktu (Fot.20).

Po wskazanym czasie przerywamy ogrzewanie, pozwalamy mieszaninie reakcyjnej ochłodzić się do kilkudziesięciu stopni, a potem mieszając zalewamy ją około 200cm3 wody destylowanej. Stały produkt odsączamy i kilkukrotnie przemywamy zimną wodą celem usunięcia rozpuszczalnych zanieczyszczeń, po czym suszymy. Tak uzyskany hydrazyd 3-nitroftalowy C8H5N3O4 ma postać brudnożółtego, bezpostaciowego proszku (Fot.21). Jego wzór strukturalny można zobaczyć na Rys.5.

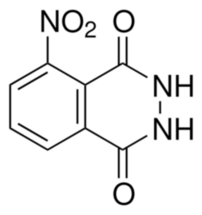
Uzyskałem 9,62g hydrazydu 3-nitroftalowego, co daje wydajność ok. 76% w przeliczeniu na wykorzystany kwas.
Etap IIIa – Dwutlenek tiomocznika
W kolejnym etapie będziemy potrzebowali środka redukującego, który pozwoli przekształcić hydrazyd 3-nitroftalowy do 3-aminoftalowego. Rolę tę może spełnić wiele substancji, takich jak np. chlorek cyny(II) SnCl2 lub ditionian(III) sodu Na2S2O4. Ja jednak chciałbym zaproponować wykorzystanie innej ciekawej substancji, jaką jest dwutlenek tiomocznika CH4N2O2S. Trzeba przyznać, że jest to nietypowy reduktor, ponieważ otrzymamy go w reakcji utleniania.
Aby wytworzyć dwutlenek tiomocznika potrzebujemy:
- tiomocznik CH4N2O - 20g
- nadtlenek wodoru H2O2 30% - 67cm3
Całą objętość nadtlenku wodoru umieszczamy w metalowym, np. aluminiowym naczyniu, które wstawiamy do łaźni zawierającej mieszaninę chłodzącą (lód z chlorkiem sodu), po czym ustawiamy na mieszadle magnetycznym (Fot.22A).

A – schłodzony nadtlenek, B – powstający biały produkt
Następnie rozpoczynamy dodawanie do mieszaniny tiomocznika, ale robimy to bardzo powoli i naprawdę niewielkimi porcjami. Reakcja ta jest ekstremalnie egzotermiczna, a powyżej 20°C zaczyna zachodzić prawie wybuchowo – dlatego kluczowe jest bardzo wydajne odprowadzanie energii cieplnej z układu reakcyjnego. Jaki będzie efekt, jeśli zaniechamy tego środka ostrożności? Można się o tym przekonać spoglądając na Fot.23 (i nagranie video, przyp.aut.). Jak widać, przy braku chłodzenia nawet w przypadku małych ilości substratów, mieszanina zaczyna bardzo gwałtownie wrzeć natychmiast po kontakcie tiomocznika z utleniaczem, a wokół rospryskuje się gorąca i silnie żrąca ciecz. Przy intensywnym chłodzeniu (<20°C), powolnym dodawaniu tiomocznika i silnym mieszaniu reakcja zachodzi spokojnie [11].

A – 5cm3 nadtlenku wodoru 30% w zlewce,
B – po dodaniu tiomocznika, widoczne gwałtowne wrzenie i rozprysk mieszaniny
W trakcie dodawania kolejnych porcji pochodnej mocznika możemy zauważyć, że z roztworu zaczyna się wytrącać białe ciało stałe (Fot.22B). Po dodaniu całości mieszanie kontynuujemy jeszcze 30 minut, a następnie osad odsączamy oraz przemywamy niewielką ilością zimnej wody, po czym suszymy i przechowujemy w ciemności.
W opisany sposób otrzymałem 20,4g dwutlenku tiomocznika, co stanowi w przybliżeniu 76% wydajności teoretycznej.
Etap IV – Od hydrazydu 3-nitroftalowego do hydrazydu 3-aminoftalowego (luminolu)
Ostatnim etapem syntezy jest redukcja grupy nitrowej hydrazydu 3-nitroftalowego do aminowej i tym samym uzyskanie luminolu. Przygotujmy więc:
- hydrazyd 3-nitroftalowy – 9,6g
- dwutlenek tiomocznika – 20,1g
Hydrazyd 3-nitroftalowy rozpuszczamy w 105cm3 wody amoniakalnej NH3(aq) o stężeniu 4,5M uzyskując ciemnoczerwony roztwór (Fot.24).

Do roztworu dodajemy następnie intensywnie mieszając dwutlenek tiomocznika. Ciecz delikatnie się ogrzewa, a jej barwa ulega rozjaśnieniu (Fot.25). Jedocześnie z roztworu uwolniają się duże ilości gazowego amoniaku, więc proces trzeba prowadzić pod wyciągiem lub na zewnątrz.

Po dodaniu całej ilości reduktora roztwór ogrzewamy do temperatury 80-90°C w czasie 30 minut, po czym chłodzimy. Ma to na celu umożliwienie reagentom jak najdokładniejsze przereagowanie.
Aby wytrącić powstały produkt musimy zobojętnić, a następnie bardzo delikatnie zakwasić roztwór kwasem chlorowodorowym. Robimy to powoli, dodając kwas małymi porcjami, ponieważ zbytnie obniżenie pH nie jest korzystne. Łatwo zauważymy moment, kiedy z roztworu wytrąci się żółty osad hydrazydu 3-aminoftalowego (Fot.26).

Osad trzeba odsączyć i kilkukrotnie przepłukać większą ilością wody destylowanej, co nie wprowadza znaczących strat z powodu bardzo małej rozpuszczalności produktu w czystej wodzie. Hydrazyd możemy oczyścić przez rekrystalizację, ale nie jest to konieczne, ponieważ jego czystość jest wystarczająca do większości zastosowań. Ma on postać żółtego proszku (Fot.27).

Otrzymałem 5,4g hydrazydu 3-aminoftalowego (luminolu) w postaci chlorowodorku C8H7N3O2·HCl (Rys.6), co stanowi 54% w przeliczeniu na wykorzystany hydrazyd 3-nitroftalowy.
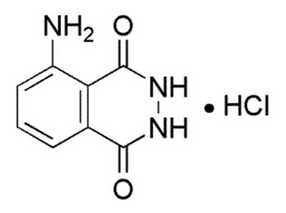
Luminol należy przechowywać w ciemności – jego trwałość jest wtedy naprawdę duża.
Chemiluminescencja luminolu
Luminol wykazuje chemiluminescencję zarówno w roztworach wodnych, jak i w wielu rozpuszczalnikach organicznych, takich jak dimetyloformamid C3H7NO oraz dimetylosulfotlenek C2H6OS. W literaturze możemy znaleźć wiele przepisów na bardzo interesujące reakcje z wykorzystaniem właściwości chemiluminescencyjnych luminolu. Aby wypróbować otrzymaną przez nas substancję proponuję wykorzystać jedną z najprostszych receptur:
| A: | 0,2g luminolu i 3g wodorotlenku sodu w 50cm3 wody destylowanej, tuż przed eksperymentem dodać 5cm3 nadtlenku wodoru 3% (aptecznej wody utlenionej) |
| B: | 0,5g heksacyjanożelazianu(III) potasu K3[Fe(CN)6] w 10cm3 wody destylowanej |
Po zmieszaniu obu roztworów jesteśmy świadkiem emisji jasnego niebieskiego światła, co - mimo dosyć szybkiego zaniku efektu - dostarcza szczególnych wrażeń.

A – roztwór chemiluminoforu i utleniacza,
B – po zmieszaniu dwóch roztworów, widoczna emisja niebieskiego światła
Na czas oraz intensywność emisji światła ma wpływ zarówno skład roztworów, jak i wykorzystany rozpuszczalnik.
Wyjaśnienie
Reakcja chemiluminescencji w tym przypadku zachodzi etapowo. W zasadowym roztworze luminol dysocjuje do postaci dwuujemnego anionu. Jest to jednak bardziej skomplikowane niż się wydaje, ponieważ ma miejsce tutaj tautomeria keto-enolowa: na drodze dysocjacji omawianego hydrazydu powstają dwie różniące się budową i rozmieszczeniem ładunku ujemnego formy: ketonowa, gdzie ładunek ujemny jest zlokalizowany na atomach azotu, oraz enolowa, w której ładunek ten jest zgromadzony na atomach tlenu. Obie formy istnieją w stanie równowagi dynamicznej, w sposób ciągły przechodząc jedna w drugą, ale dalszym reakcjom ulega forma enolowa, jako mniej stabilna chemicznie. Zostaje ona utleniona przez nadtlenek wodoru, co jest katalizowane dodatkiem heksacyjanożelazianu(III) potasu. Produktem jest w tym przypadku cykliczny nadtlenek. Z racji istnienia w jego strukturze mostka nadtlenkowego związek ten jest bardzo nietwały. Dochodzi do spontanicznego rozpadu, czego produktem jest cząsteczka azotu oraz 3-aminoftalan w stanie wzbudzonym, który z punktu widzenia termodynamiki jest niestabilny, a więc dochodzi do spontanicznego przejścia do stanu podstawowego. Zgodnie z zasadą zachowania nadwyżka energii zostaje wypromieniowana do środowiska pod postacią promieniowania elektromagnetycznego, w tym przypadku o długości fali odpowiadającej światłu niebieskiemu.
Luminol znajduje zastosowanie także poza dydaktyką, np. w kryminalistyce do wstępnego wykrywania śladów krwi na miejscu zbrodni, ale też m.in. w analizie chemicznej.
Literatura:
- [1] Ples M., Blask w kolbie - synteza i chemiluminescencja lucygeniny, Chemia w Szkole, 3 (2023), Agencja AS Józef Szewczyk, str. 44-50 powrót
- [2] Ples M., Fosfor - alchemiczne światło, Chemia w Szkole, 2 (2019), Agencja AS Józef Szewczyk, str. 13-17 powrót
- [3] Ples M., Lofina - wielka synteza, Chemia w Szkole, 6 (2022), Agencja AS Józef Szewczyk, str. 43-50 powrót
- [4] Ples M., Co i jak można otrzymać z piasku? Nieznane oblicze krzemu, Chemia w Szkole, 6 (2016), Agencja AS Józef Szewczyk, str. 38-43 powrót
- [5] Haynes W. M. (red.), CRC Handbook of Chemistry and Physics (wyd. 95), CRC Press, 2014, str. 3-18 powrót
- [6] Sheikh I. A., Stereoselectivity and the potential endocrine disrupting activity of di-(2-ethylhexyl)phthalate (DEHP) against human progesterone receptor: a computational perspective, Journal of applied toxicology, 36 (5), 2016, str. 741-747 powrót
- [7] Hassa R., Mrzigod J., Podręczny słownik chemiczny, Videograf II, Katowice, 2004, str. 167 powrót
- [8] Greenwood N. N., Earnshaw A., Chemistry of the elements, Pergamon Press, 1984, str. 427-428 powrót
- [9] Hofmann A. W., Ueber die Einwirkung des Broms in alkalischer Lösung auf Amide, Berichte der Deutschen Chemischen Gesellschaft, 14, 1881, str. 2725-2736 powrót
- [10] Wallis E. S., Lane J. F., The Hofmann Reaction, Organic Reactions, 3, 1949, str. 267-306 powrót
- [11] Pluciński T., Synteza luminolu, dostępne online: http://www.tomek.strony.ug.edu.pl/syntezaluminolu.htm [dostęp 14.10.2023] powrót
Wszystkie fotografie i rysunki zostały wykonane przez autora
Uzupełnienie autora
Chemiluminescencja luminolu jest tak efektownym zjawiskiem, że warto je wykorzystywać w czasie zajęć np. do uatrakcyjnienia przekazywania treści związanych z przemianami energetycznymi na poziomie molekularnym.
Dosyć mało znana i rzadko wykorzystywana zdaje się możliwość transferu energii w czasie chemiluminescencji luminolu na obecne w roztworze cząsteczki barwnika fluorescencyjnego, np. fluoresceiny C20H12O5. Barwa emitowanego światła zmienia się wtedy na charakterystyczną dla tej substancji.

Co ciekawe, katalizatorem reakcji utleniania luminolu mogą być także związki innych metali, np. miedzi Cu.

Możliwe jest także wykorzystanie luminolu jako chemiluminescencyjnego wskaźnika w reakcjach oscylacyjnych.
Przykłady te oczywiście nie wyczerpują możliwości związanych z edukacyjnym i naukowym wykorzystaniem luminolu, zachęcam więc do samodzielnych eksperymentów.
Marek Ples