Fluorescencyjna termochromia - zimno, ale nie bezbarwnie
Poniższy artykuł został opublikowany pierwotnie w czasopiśmie dla nauczycieli Chemia w Szkole (4/2017):

Niska temperatura kojarzy się nam zwykle z ubóstwem barw. Łatwo to zrozumieć pamiętając o naturalnym skojarzeniu z zimą, jako porą roku odznaczającą się na naszej szerokości geograficznej przerwą w wegetacji i bielą śniegu. Udowodnię jednak, że nawet bardzo niskie temperatury niekoniecznie zmuszają nas do porzucenia piękna i bogactwa barw.
W tym miejscu chciałbym przypomnieć Szanownemu Czytelnikowi o zjawiskach zbiorczo nazywanych terminem chromotropizmów. Polegają one na odwracalnej zmianie barwy związków chemicznych pod wpływem zmiany warunków fizycznych lub chemicznych [1]. Barwa może zmieniać się w wyniku działania różnych czynników, np. zmian ciśnienia (piezochromizm), oświetlenia (fotochromizm), pola magnetycznego (magnetochromizm), rodzaju rozpuszczalnika (solwatochromizm).
Jednym z szerzej znanych przykładów chromotropizmów jest termochromizm, nazywany też termochromią. Polega on na zależności zmiany barwy substancji od temperatury. Takie właściwości wykazuje np. jodek rtęci HgI2 – w temperaturze pokojowej jest czerwony, natomiast po ogrzaniu do 126°C staje się żółty - cały proces jest odwracalny [2]. Chociaż doświadczenia z tą substancją są interesujące, to mają jednak dosyć poważną z punktu widzenia eksperymentatora wadę: związki rtęci są silnie trujące, a ich odpowiednie unieszkodliwienie i składowanie jest dosyć trudne. Bezpieczniejszym przykładem substancji wykazujących tego rodzaju właściwości jest - biały w niezbyt wysokich temperaturach - tlenek cynku ZnO (Fot.1A), który po ogrzaniu odwracalnie zmienia barwę na żółtą (Fot.1B). Jest to w tym przypadku spowodowane powstawaniem tzw. centrów barwnych (centrów F) – defektów sieci krystalicznej, w których miejsce jonu ujemnego zostaje zajęte przez jeden lub więcej niesparowanych elektronów [3].

Oczywiście w przypadku termochromii dochodzi do zmiany barwy światła odbitego od danej substancji. Istnieje jednak pewne interesujące zjawisko, w którym zmienia się barwa światła emitowanego przez substancję – dlatego bywa ono nazywane fluorescencyjną termochromią. Fenomen ten występuje bardzo wyraźnie dla pewnego łatwego w syntezie związku, tj. jodku monopirydynamiedzi(I) [4]. W razie problemów ze zdobyciem potrzebnych substancji opisuję też wersję doświadczenia, którą można wykonać z wykorzystaniem bardzo łatwych do zdobycia, tanich i bezpiecznych substancji.
Potrzebne substancje
Chcąc otrzymać potrzebny nam do doświadczenia kompleks pirydyny i jodku miedzi musimy zgromadzić:
- pirydynę C5H5N
- jodek miedzi(I) CuI
Jodek miedzi(I) ma działanie toksyczne i drażniące. Pirydyna wykazuje szkodliwe własności w przypadku połknięcia, inhalacji lub kontaktu ze skórą. Prawdopodobne jest także jej działanie rakotwórcze. Ciecz ta jest lotna i ma odrażający zapach.
Jodek miedzi(I) może okazać się trudnodostępny. Można go jednak łatwo otrzymać w reakcji jodku potasu z rozpuszczalną solą miedzi(II) - na przykład z siarczanem(VI) miedzi(II) CuSO4. Po zmieszaniu wodnych roztworów obu soli dochodzi do reakcji opisanej poniższym równaniem:
Obok siarczanu(VI) potasu K2SO4 powstaje wtedy jodek miedzi(II) CuI2. My jednak przecież potrzebujemy jodku, w którym miedź wykazywałaby wartościowość równą I. Nic nie stoi na przeszkodzie, ponieważ uzyskany jodek miedzi(II) jest nietrwały i ulega natychmiastowej reakcji dysproporcjonowania do jodku miedzi(I) oraz pierwiastkowego jodu I2:
Powstały osad jodku miedzi(I) należy odsączyć (najlepiej próżniowo), a następnie kilkukrotnie przemyć zimnym etanolem w celu usunięcia pozostałości jodu, który w razie potrzeby można odzyskać z przesączu.
Otrzymany jodek miedzi(I) ma postać białego proszku (Fot.2).

Drugim wymaganym reagentem jest pirydyna. Jest to organiczny aromatyczny związek heterocykliczny. Oznacza to, że w pierścieniu jego cząsteczki występuje przynajmniej jeden atom pierwiastka innego niż węgiel – w tym przypadku jest to azot N. Doskonale ukazuje to wzór strukturalny pirydyny (Rys.1).
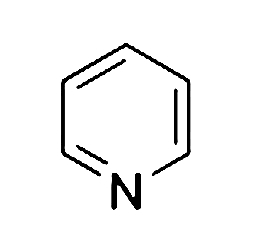
Substancja ta jest bezbarwną cieczą o gęstości nieco mniejszej od gęstości wody (Fot.3).

Pirydynę należy przechowywać w szczelnie zamkniętych naczyniach z powodu jej lotności i toksyczności.
Otrzymywanie kompleksu
By wytworzyć odpowiedni kompleks, wystarczy dodać kilka miligramów jodku miedzi(I) do niewielkiej objętości (około 5 cm3) pirydyny. Po wstrząśnięciu jodek roztworzy się, a roztwór przybierze barwę żółtawą (Fot.4).

W ten sposób uzyskaliśmy roztwór kompleksu jodku tripirydynamiedzi(I) (C5H5N)3CuI w pirydynie.
Roztworem tym należy nasączyć paski papieru; nie można tu użyć żadnego papieru zawierającego wybielacze optyczne. Dlatego na pewno nie nada się tutaj papier do drukarki ani papier zeszytowy. Najlepszy będzie papier filtracyjny pozyskany ze zwykłego sączka.
Papier należy następnie ostrożnie wysuszyć w temperaturze 40-60°C. Najpierw odparowuje wtedy nadmiar pirydyny, po czym kompleks zaczyna odłączać od siebie jej cząsteczki. Po odszczepieniu jednej przechodzi w jodek dipirydynamiedzi(I) (C5H5N)2CuI, wykazujący niezbyt silną bladozieloną fluorescencję pod wpływem światła ultrafioletowego. Przy dalszym ogrzewaniu kompleks odszczepia kolejną cząsteczkę pirydyny i na papierze pozostaje warstewka stabilnego w temperaturze pokojowej jodku monopirydynamiedzi(I) (C5H5N)CuI. I to właśnie ten związek jest głównym przedmiotem naszego zainteresowania!
Uwaga praktyczna: ogrzewania nie należy prowadzić w zbyt wysokiej temperaturze ani przesadnie długo, ponieważ wtedy potrzebny kompleks może ulec rozkładowi do jodku i pirydyny.
Fluorescencyjna termochromia
Spójrzmy na Fot.5 - pasek papieru B został pokryty kompleksem, w przeciwieństwie do paska A.

W świetle widzialnym oba fragmenty papieru wyglądają tak samo; nie udaje się zaobserwować żadnej różnicy w barwie.
Użycie światła ultrafioletowego ujawnia zdecydowanie odmienny widok (Fot.6).

Pasek A pozostaje w dalszym ciągu ciemny, ale pokryty omawianym kompleksem pasek B fluoryzuje pięknym jasnym światłem koloru cytrynowo-żółtego!
Sprawdźmy teraz co się stanie po schłodzeniu substancji w ciekłym azocie o temperaturze wrzenia równej -196°C. Przy pracy z tą substancją trzeba oczywiście zachować daleko idącą ostrożność, ponieważ tak niska temperatura może z łatwością spowodować niebezpieczne odmrożenia. Przy krótkotrwałym kontakcie ciekłego azotu ze skórą naszym sprzymierzeńcem jest jednak zjawisko Leidenfrosta – ciecz gwałtownie wrząc tworzy izolacyjną warstwę gazu, która przez krótki czas zabezpiecza przed urazem termicznym [5].
Fluorescencja w dalszym ciągu jest wyraźnie widoczna, ale zachodzi zaskakująca zmiana: w niskiej temperaturze miejsce barwy żółtej zajmuje fioletowo-niebieska (Fot.7).

Po ogrzaniu fluorescencja na powrót przyjmuje barwę żółtą (Fot.8).

Różnice w barwie można porównać analizując Fot.9.

Przejście barwne jest bardzo kontrastowe; podczas ogrzewania paska papieru gradient temperatury jest widoczny jako przemieszczająca się granica między barwą fioletowo-niebieską a żółtą (Fot.10).

Jeszcze prościej
Jeśli nie mamy dostępu do substancji potrzebnych do wykonania uprzednio opisanych doświadczeń to nie trzeba się załamywać, ponieważ przygotowałem także inne ich warianty. Różnica barw nie jest w tym przypadku aż tak kontrastowa, ale wymagane materiały są tanie i łatwe do zdobycia:
- glicerol C3H8O3
- mleczan etakrydyny C18H21N3O4
Zarówno glicerol (glicerynę), jak i mleczan etakrydyny możemy z nabyć w aptece. Ten drugi, pod handlową nazwą riwanol lub akrinol znalazł zastosowanie jako środek dezynfekujący.
Chociaż riwanol nie jest uważany za silnie toksyczny, to w dużych ilościach może wykazywać działania niepożądane. W stanie wilgotnym pozostawia na skórze trudne do usunięcia żółte plamy.
Riwanol zarówno w stanie stałym, jak i w roztworze wodnym wykazuje bardzo silną fluorescencję. Jest ona zauważalna gołym okiem nawet dla stężeń dochodzących do jednej milionowej części procenta [6].
Aby wykonać doświadczenia jest konieczne przygotowanie roztworu riwanolu w glicerolu. Roztwór nie może być zbyt stężony – wystarczy w kilku-kilkunastu centymetrach sześciennych cieczy rozpuścić dosłownie szczyptę barwnika!
Rozpuszczanie substancji stałej w glicerolu jest dosyć powolne. Silnie mieszanie wydatnie przyspiesza ten proces.
Glicerolowy roztwór riwanolu (podobnie jak wodny) wykazuje bardzo silną fluorescencję pod wpływem światła ultrafioletowego (Fot.11A). Zauważmy, że jej barwa jest intensywnie jasnozielona.

Tak jest jednak jedynie w temperaturze pokojowej. Aby przekonać się, jaki efekt spowoduje ochłodzenie roztworu, w tym przypadku wystarczy nam suchy lód, tj. zestalony dwutlenek węgla CO2. Sublimując zachowuje on temperaturę −78,5°C i dzięki zastosowaniu w chłodnictwie bywa łatwiej dostępny niż ciekły azot. Pamiętać należy jednak, że mimo temperatury dużo wyższej niż w przypadku ciekłego azotu, suchy lód w dalszym ciągu może spowodować dotkliwe i trudno gojące się odmrożenia – z tych powodów trzeba zachować ostrożność.
Po ochłodzeniu suchym lodem roztwór w zauważalny sposób zwiększa swoją lepkość, zaś barwa jego fluorescencji zmienia się na wyraźnie niebiesko-zieloną (Fot.11B). Po ogrzaniu następuje powrót do jasnozielonej barwy fluorescencji.
W tym przypadku możemy zaobserwować także inne interesujące zjawisko – fosforescencję. Uprzedzam jednak, że przy wykorzystaniu suchego lodu intensywność i długość trwania poświaty są stosunkowo małe – zjawisko zaczyna być łatwe do zauważenia dopiero po ochłodzeniu roztworu ciekłym azotem. Po naświetleniu zawartości naczynia i wyłączeniu źródła światła UV można zaobserwować jasną, trwającą pewien czas żółtą fosforescencję roztworu (Fot.12).

Przechowywanie
Glicerolowy roztwór riwanolu i otrzymane uprzednio impregnowane kompleksem jodku monopirydynamiedzi(I) paski bibuły są trwałe i można je wykorzystywać wielokrotnie. Należy je jednak przechowywać w odpowiednich warunkach, tj. bez dostępu wilgoci i światła.
Wyjaśnienie
Wiemy, że każde ciało podgrzane do odpowiednio wysokiej temperatury zaczyna świecić, podobnie jak rozgrzany w płomieniu palnika pręt metaliczny. Należy jednak pamiętać, że materia wysyła promieniowanie elektromagnetyczne w każdej temperaturze wyższej od zera bezwzględnego - zaczyna być ono dostrzegalne dla nas dopiero w odpowiednio wysokiej temperaturze. Promieniowanie to jest efektem tak zwanej emisji termicznej.
Odrębnym ze względu na mechanizm jego powstawania zjawiskiem jest luminescencja, w trakcie której promieniowanie elektromagnetyczne z zakresu światła widzialnego jest emitowane z przyczyn innych niż wysoka temperatura. Istnieje wiele typów luminescencji, różniących się czynnikiem pobudzającym dane ciało do świecenia. Może to być przepływ prądu elektrycznego (elektroluminescencja), pewne reakcje chemiczne (chemiluminescencja), kruszenie kryształów (tryboluminescencja) i inne.
Fluorescencja jest także jednym z rodzajów luminescencji (fotoluminescencji). Jest to zjawisko emitowania światła przez atom lub cząsteczkę po pobudzeniu światłem, najczęściej promieniowaniem UV. Zjawisko uznaje się za fluorescencję, gdy po ustaniu promieniowania wzbudzającego następuje szybki zanik emisji, tzn. w czasie do 10−8s.
Zrozumienie zachodzących tu procesów może ułatwić wykorzystanie tzw. diagramów Jabłońskiego. Obrazują one uproszczony obraz względnego rozmieszczenia poziomów energetycznych cząsteczki. Odpowiedni w tym przypadku diagram został przedstawiony na Rys.2.
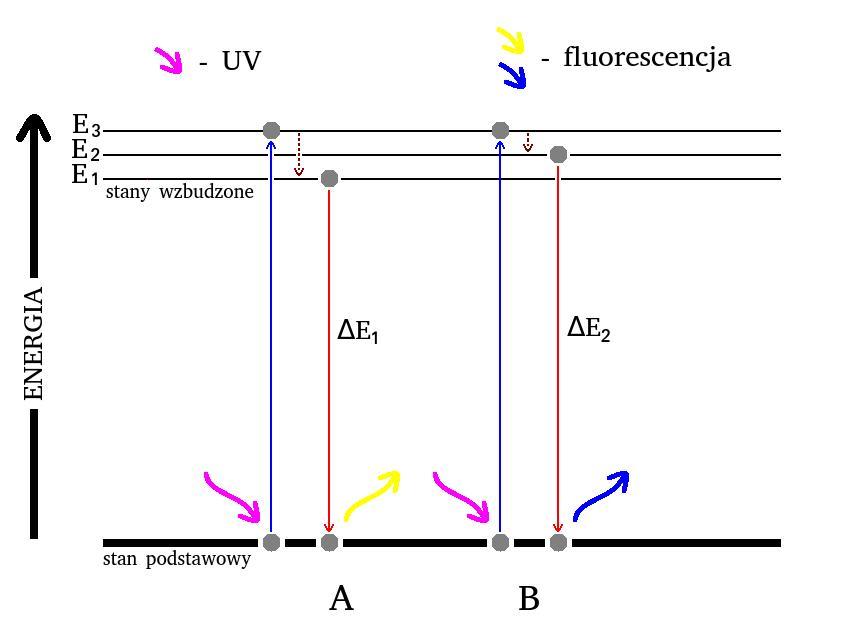
W normalnych warunkach cząstka przez większość czasu znajduje się w stanie podstawowym, o najniższej energii. Stan ten jest stabilny. Po pochłonięciu odpowiedniej porcji energii (tutaj w postaci światła ultrafioletowego UV) następuje przejście do stanu wzbudzonego o wyższej energii – tutaj E3.
Część energii wzbudzenia zostaje jednak prawie natychmiast rozproszona, np. na skutek przejść oscylacyjno-rotacyjnych (ang. vibrational-rotational, VR). Z tego powodu cząstka przechodzi na niższy poziom energetyczny, który w odniesieniu do stanu podstawowego jest w dalszym ciągu stanem wzbudzonym.
Wiemy, że cząstki w temperaturach wyższych od zera absolutnego intensywnie drgają. Amplituda owych drgań rośnie wraz z temperaturą. Z tego powodu w wyższej temperaturze (Rys.2A) podczas przejścia oscylacyjno-rotacyjnego VR zostanie rozproszona większa ilość energii niż w niższej temperaturze (Rys.2B). Dlatego w wyniku VR cząstka w pierwszym przypadku przyjmie niższy stan energetyczny (E1) niż w drugim (E2).
Natura dąży do równowagi - stabilny jest stan o możliwie najniższej energii. Dlatego (w przypadku fluorescencji) bardzo szybko zachodzi przeskok do stanu podstawowego. Różnica energii ΔE zgodnie z zasadą zachowania energii zostaje oddana do środowiska w postaci promieniowania elektromagnetycznego o odpowiedniej długości fali [7].
Podczas przeskoku do stanu podstawowego różnica energii między stanem wzbudzonym, a podstawowym ΔE1 jest mniejsza niż ΔE2. Długość fali wypromieniowanego światła (jego barwa) jest ściśle związana z jego energią. W przypadku jodku monopirydynamiedzi(I) różnica energii w warunkach temperatury pokojowej (ΔE1) odpowiada światłu żółtemu, zaś po schłodzeniu (ΔE2) fioletowo-niebieskiemu. Analogicznie wygląda sytuacja w przypadku glicerolowego roztworu riwanolu.
Widzimy jasno, że z powodu rozpraszania energii promieniowanie oddane ma zawsze niższą energię niż promieniowanie wzbudzające. Zjawisko to nazywamy przesunięciem Stokesa i mogliśmy je zaobserwować dla obu substancji w każdej temperaturze. Zauważmy jednak, że w opisywanych doświadczeniach zmienialiśmy wielkość przesunięcia Stokesa za pomocą temperatury substancji.
Jeśli chodzi o fosforescencję schłodzonego roztwory rywanolu, to kluczową kwestią jest tutaj fakt, że w tym przypadku stan wzbudzony możemy określić jako metastabilny. Cząstka może w nim pozostać przez dużo dłuższy czas po ustaniu czynnika wzbudzającego niż ma to miejsce w fluorescencji. W substancjach wykazujących fosforescencję dochodzi do stosunkowo częstych przejść międzysystemowych między stanem singletowym a trypletowym. Najbardziej prawdopodobne są jednak przejścia między stanami o tej samej multipletowości – mówi o tym kwantowa reguła wyboru. Przejście do stanu podstawowego jest więc utrudnione z powodu niewielkiego prawdopodobieństwa takiego procesu, przez co uwalnianie energii wzbudzenia trwa na tyle długo, że możemy je zaobserwować jako powoli zanikającą poświatę. Wiele substancji wykazuje podobne właściwości, np. domieszkowany manganem siarczek cynku ZnS:Mn [8].
W ten stosunkowo prosty sposób udało nam się wniknąć w mechanizmy przemian energetycznych zachodzących w skali atomowej. Trzeba także zaznaczyć, że tego typu właściwości nie są jedynie ciekawostką; znajdują one zastosowanie w wielu dziedzinach współczesnej nauki.
Literatura:
- [1] Linerta W., Fukudab Y., Camarda A., Chromotropism of coordination compounds and its applications in solution, Coordination Chemistry Reviews, 218, 2001 powrót
- [2] Pluciński T., Doświadczenia chemiczne, Wydawnictwo Adamantan, Warszawa, 1997, str. 212-213 powrót
- [3] Schulman J. H., Compton W.D., Color Centers in Solids, Pergamon, Oxford, 1962 powrót
- [4] Parmeggiani F., Sacchetti A., Preparation and Luminescence Thermochromism Copper(I)-Pyridine-Iodide Clusters, Journal of Chemical Education, 89 (2012), str. 946-949 powrót
- [5] Szczeniowski S., Fizyka doświadczalna - Ciepło i fizyka molekularna, Państwowe Wydawnictwo Naukowe, Warszawa, 1953 powrót
- [6] Ples M., Więcej światła! O fluorescencji riwanolu, Chemia w Szkole, 6 (2015), Agencja AS Józef Szewczyk, str. 16-18 powrót
- [7] Valeur B., Berberan-Santos M. R. N., Molecular Fluorescence: Principles and Applications, Wiley-VCH, 2012, str. 64 powrót
- [8] Ples M., Jak uwięzić światło? O skutkach domieszkowania siarczku cynku, Chemia w Szkole, 1 (2017), Agencja AS Józef Szewczyk, str. 12-18 powrót
Wszystkie fotografie wykonane przez autora
Uzupełnienie autora
Jako dodatek do powyższego tekstu proponuję animację ukazującą w czasie rzeczywistym zmianę barw omawianego w artykule kompleksu miedzi przy ogrzewaniu po uprzednim zanurzeniu w ciekłym azocie:

Marek Ples